Co-reporter:Akifumi Kato, Mitsuo Kuratani, Tatsuo Yanagisawa, Kazumasa Ohtake, Akiko Hayashi, Yoshimi Amano, Kaname Kimura, Shigeyuki Yokoyama, Kensaku Sakamoto, and Yasuhisa Shiraishi
Bioconjugate Chemistry August 16, 2017 Volume 28(Issue 8) pp:2099-2099
Publication Date(Web):July 20, 2017
DOI:10.1021/acs.bioconjchem.7b00265
The site-specific chemical conjugation of proteins, following synthesis with an expanded genetic code, promises to advance antibody-based technologies, including antibody drug conjugation and the creation of bispecific Fab dimers. The incorporation of non-natural amino acids into antibodies not only guarantees site specificity but also allows the use of bio-orthogonal chemistry. However, the efficiency of amino acid incorporation fluctuates significantly among different sites, thereby hampering the identification of useful conjugation sites. In this study, we applied the codon reassignment technology to achieve the robust and efficient synthesis of chemically functionalized antibodies containing Nε-(o-azidobenzyloxycarbonyl)-l-lysine (o-Az-Z-Lys) at defined positions. This lysine derivative has a bio-orthogonally reactive group at the end of a long side chain, enabling identification of multiple new positions in Fab-constant domains, allowing chemical conjugation with high efficiency. An X-ray crystallographic study of a Fab variant with o-Az-Z-Lys revealed high-level exposure of the azido group to solvent, with six of the identified positions subsequently used to engineer “Variabodies”, a novel antibody format allowing various connections between two Fab molecules. Our findings indicated that some of the created Variabodies exhibited agonistic activity in cultured cells as opposed to the antagonistic nature of antibodies. These results showed that our approach greatly enhanced the availability of antibodies for chemical conjugation and might aid in the development of new therapeutic antibodies.
Co-reporter:Haruhiko Ehara, Takashi Umehara, Shun-ichi Sekine, Shigeyuki Yokoyama
Biochemical and Biophysical Research Communications 2017 Volume 487, Issue 2(Issue 2) pp:
Publication Date(Web):27 May 2017
DOI:10.1016/j.bbrc.2017.04.039
•K. pastoris Pol II was crystallized in three different crystal forms.•One of the structures showed a highly-closed conformation of the stalk and the clamp.•Structural comparisons revealed the ranges of the stalk and clamp movements of Pol II.RNA polymerase II (Pol II) is a 12-subunit protein complex that conducts the transcription of mRNA and some small RNAs. In this work, the crystal structure of Pol II from the methylotropic yeast Komagataella pastoris (Pichia pastoris) was determined. While the structure is highly homologous to that of Pol II from the budding yeast Saccharomyces cerevisiae, the stalk and clamp modules of the K. pastoris Pol II displayed large inward rotations, closing the central cleft to a greater extent than in the known S. cerevisiae Pol II structures. The conformational differences reflect the inherent flexibilities of the stalk and the clamp, as additional low-resolution structures of K. pastoris Pol II in different crystal forms revealed diverse stalk and clamp orientations. Comparisons with other eukaryotic/archaeal RNA polymerase structures in the Protein Data Bank revealed the distributions of the stalk and clamp orientations. The conformational plasticity should be essential for transcriptional functions and binding various regulatory factors.
Co-reporter:Yong-Tae Kim;Hongfei Wang;Tomonari Muramatsu;Chie Takemoto;Mikako Shirouzu;Wataru Nishii;Takaho Terada
PNAS 2016 Volume 113 (Issue 46 ) pp:12997-13002
Publication Date(Web):2016-11-15
DOI:10.1073/pnas.1601327113
The 3C-like protease (3CLpro) of severe acute respiratory syndrome coronavirus (SARS-CoV) cleaves 11 sites in the polyproteins, including its own N- and
C-terminal autoprocessing sites, by recognizing P4–P1 and P1′. In this study, we determined the crystal structure of 3CLpro with the C-terminal prosequence and the catalytic-site C145A mutation, in which the enzyme binds the C-terminal prosequence
of another molecule. Surprisingly, Phe at the P3′ position [Phe(P3′)] is snugly accommodated in the S3′ pocket. Mutations
of Phe(P3′) impaired the C-terminal autoprocessing, but did not affect N-terminal autoprocessing. This difference was ascribed
to the P2 residue, Phe(P2) and Leu(P2), in the C- and N-terminal sites, as follows. The S3′ subsite is formed by Phe(P2)-induced
conformational changes of 3CLpro and the direct involvement of Phe(P2) itself. In contrast, the N-terminal prosequence with Leu(P2) does not cause such conformational
changes for the S3′ subsite formation. In fact, the mutation of Phe(P2) to Leu in the C-terminal autoprocessing site abolishes
the dependence on Phe(P3′). These mechanisms explain why Phe is required at the P3' position when the P2 position is occupied
by Phe rather than Leu, which reveals a type of subsite cooperativity. Moreover, the peptide consisting of P4–P1 with Leu(P2)
inhibits protease activity, whereas that with Phe(P2) exhibits a much smaller inhibitory effect, because Phe(P3′) is missing.
Thus, this subsite cooperativity likely exists to avoid the autoinhibition of the enzyme by its mature C-terminal sequence,
and to retain the efficient C-terminal autoprocessing by the use of Phe(P2).
Co-reporter:Shigeyuki Yokoyama;Kei Yura
Journal of Structural and Functional Genomics 2016 Volume 17( Issue 4) pp:67
Publication Date(Web):2016 December
DOI:10.1007/s10969-016-9213-1
Co-reporter:Kazuhiro Kashiwagi;Tomoaki Shigeta
Journal of Structural and Functional Genomics 2016 Volume 17( Issue 1) pp:33-38
Publication Date(Web):2016 March
DOI:10.1007/s10969-016-9203-3
Tight control of protein synthesis is necessary for cells to respond and adapt to environmental changes rapidly. Eukaryotic translation initiation factor (eIF) 2B, the guanine nucleotide exchange factor for eIF2, is a key target of translation control at the initiation step. The nucleotide exchange activity of eIF2B is inhibited by the stress-induced phosphorylation of eIF2. As a result, the level of active GTP-bound eIF2 is lowered, and protein synthesis is attenuated. eIF2B is a large multi-subunit complex composed of five different subunits, and all five of the subunits are the gene products responsible for the neurodegenerative disease, leukoencephalopathy with vanishing white matter. However, the overall structure of eIF2B has remained unresolved, due to the difficulty in preparing a sufficient amount of the eIF2B complex. To overcome this problem, we established the recombinant expression and purification method for eIF2B from the fission yeast Schizosaccharomyces pombe. All five of the eIF2B subunits were co-expressed and reconstructed into the complex in Escherichia coli cells. The complex was successfully purified with a high yield. This recombinant eIF2B complex contains each subunit in an equimolar ratio, and the size exclusion chromatography analysis suggests it forms a heterodecamer, consistent with recent reports. This eIF2B increased protein synthesis in the reconstituted in vitro human translation system. In addition, disease-linked mutations led to subunit dissociation. Furthermore, we crystallized this functional recombinant eIF2B, and the crystals diffracted to 3.0 Å resolution.
Co-reporter:Naoya Tochio;Takashi Umehara
Journal of Structural and Functional Genomics 2015 Volume 16( Issue 2) pp:55-65
Publication Date(Web):2015 June
DOI:10.1007/s10969-015-9196-3
ZFAT is a transcriptional regulator, containing eighteen C2H2-type zinc-fingers and one AT-hook, involved in autoimmune thyroid disease, apoptosis, and immune-related cell survival. We determined the solution structures of the thirteen individual ZFAT zinc-fingers (ZF) and the tandemly arrayed zinc-fingers in the regions from ZF2 to ZF5, by NMR spectroscopy. ZFAT has eight uncommon bulged-out helix-containing zinc-fingers, and six of their structures (ZF4, ZF5, ZF6, ZF10, ZF11, and ZF13) were determined. The distribution patterns of the putative DNA-binding surface residues are different among the ZFAT zinc-fingers, suggesting the distinct DNA sequence preferences of the N-terminal and C-terminal zinc-fingers. Since ZFAT has three to five consecutive tandem zinc-fingers, which may cooperatively function as a unit, we also determined two tandemly arrayed zinc-finger structures, between ZF2 to ZF4 and ZF3 to ZF5. Our NMR spectroscopic analysis detected the interaction between ZF4 and ZF5, which are connected by an uncommon linker sequence, KKIK. The ZF4–ZF5 linker restrained the relative structural space between the two zinc-fingers in solution, unlike the other linker regions with determined structures, suggesting the involvement of the ZF4–ZF5 interfinger linker in the regulation of ZFAT function.
Co-reporter:Isao Masuda;Takuhiro Ito;Se Won Suh;Ken-ichi Yoshida;Ya-Ming Hou;Shun-ichi Sekine;Sakurako Goto-Ito
PNAS 2015 Volume 112 (Issue 31 ) pp:E4197-E4205
Publication Date(Web):2015-08-04
DOI:10.1073/pnas.1422981112
The deep trefoil knot architecture is unique to the SpoU and tRNA methyltransferase D (TrmD) (SPOUT) family of methyltransferases
(MTases) in all three domains of life. In bacteria, TrmD catalyzes the N1-methylguanosine (m1G) modification at position 37 in transfer RNAs (tRNAs) with the 36GG37 sequence, using S-adenosyl-l-methionine (AdoMet) as the methyl donor. The m1G37-modified tRNA functions properly to prevent +1 frameshift errors on the ribosome. Here we report the crystal structure
of the TrmD homodimer in complex with a substrate tRNA and an AdoMet analog. Our structural analysis revealed the mechanism
by which TrmD binds the substrate tRNA in an AdoMet-dependent manner. The trefoil-knot center, which is structurally conserved
among SPOUT MTases, accommodates the adenosine moiety of AdoMet by loosening/retightening of the knot. The TrmD-specific regions
surrounding the trefoil knot recognize the methionine moiety of AdoMet, and thereby establish the entire TrmD structure for
global interactions with tRNA and sequential and specific accommodations of G37 and G36, resulting in the synthesis of m1G37-tRNA.
Co-reporter:Tatsuo Yanagisawa;Ryohei Ishii
Journal of Structural and Functional Genomics 2015 Volume 16( Issue 1) pp:25-41
Publication Date(Web):2015 March
DOI:10.1007/s10969-015-9193-6
The putative translation elongation factor Mbar_A0971 from the methanogenic archaeon Methanosarcina barkeri was proposed to be the pyrrolysine-specific paralogue of EF-Tu (“EF-Pyl”). In the present study, the crystal structures of its homologue from Methanosarcina mazei (MM1309) were determined in the GMPPNP-bound, GDP-bound, and apo forms, by the single-wavelength anomalous dispersion phasing method. The three MM1309 structures are quite similar (r.m.s.d. < 0.1 Å). The three domains, corresponding to domains 1, 2, and 3 of EF-Tu/SelB/aIF2γ, are packed against one another to form a closed architecture. The MM1309 structures resemble those of bacterial/archaeal SelB, bacterial EF-Tu in the GTP-bound form, and archaeal initiation factor aIF2γ, in this order. The GMPPNP and GDP molecules are visible in their co-crystal structures. Isothermal titration calorimetry measurements of MM1309·GTP·Mg2+, MM1309·GDP·Mg2+, and MM1309·GMPPNP·Mg2+ provided dissociation constants of 0.43, 26.2, and 222.2 μM, respectively. Therefore, the affinities of MM1309 for GTP and GDP are similar to those of SelB rather than those of EF-Tu. Furthermore, the switch I and II regions of MM1309 are involved in domain–domain interactions, rather than nucleotide binding. The putative binding pocket for the aminoacyl moiety on MM1309 is too small to accommodate the pyrrolysyl moiety, based on a comparison of the present MM1309 structures with that of the EF-Tu·GMPPNP·aminoacyl-tRNA ternary complex. A hydrolysis protection assay revealed that MM1309 binds cysteinyl (Cys)-tRNACys and protects the aminoacyl bond from non-enzymatic hydrolysis. Therefore, we propose that MM1309 functions as either a guardian protein that protects the Cys moiety from oxidation or an alternative translation factor for Cys-tRNACys.
Co-reporter:Hiroaki Tanabe;Kanna Motoyama;Mariko Ikeda
Journal of Structural and Functional Genomics 2015 Volume 16( Issue 1) pp:11-23
Publication Date(Web):2015 March
DOI:10.1007/s10969-014-9192-z
The adiponectin receptors (AdipoR1 and AdipoR2) are membrane proteins with seven transmembrane helices. These receptors regulate glucose and fatty acid metabolism, thereby ameliorating type 2 diabetes. The full-length human AdipoR1 and a series of N-terminally truncated mutants of human AdipoR1 and AdipoR2 were expressed in insect cells. In small-scale size exclusion chromatography, the truncated mutants AdipoR1Δ88 (residues 89–375) and AdipoR2Δ99 (residues 100–386) eluted mostly in the intact monodisperse state, while the others eluted primarily as aggregates. However, gel filtration chromatography of the large-scale preparation of the tag-affinity-purified AdipoR1Δ88 revealed the presence of an excessive amount of the aggregated state over the intact state. Since aggregation due to contaminating nucleic acids may have occurred during the sample concentration step, anion-exchange column chromatography was performed immediately after affinity chromatography, to separate the intact AdipoR1Δ88 from the aggregating species. The separated intact AdipoR1Δ88 did not undergo further aggregation, and was successfully purified to homogeneity by gel filtration chromatography. The purified AdipoR1Δ88 and AdipoR2Δ99 proteins were characterized by thermostability assays with 7-diethylamino-3-(4-maleimidophenyl)-4-methyl coumarin, thin layer chromatography of bound lipids, and surface plasmon resonance analysis of ligand binding, demonstrating their structural integrities. The AdipoR1Δ88 and AdipoR2Δ99 proteins were crystallized with the anti-AdipoR1 monoclonal antibody Fv fragment, by the lipidic mesophase method. X-ray diffraction data sets were obtained at resolutions of 2.8 and 2.4 Å, respectively.
Co-reporter:Dr. Tatsuo Yanagisawa;Mihoko Takahashi;Dr. Takahito Mukai;Shin Sato;Masatoshi Wakamori;Dr. Mikako Shirouzu;Dr. Kensaku Sakamoto;Dr. Takashi Umehara; Dr. Shigeyuki Yokoyama
ChemBioChem 2014 Volume 15( Issue 12) pp:
Publication Date(Web):
DOI:10.1002/cbic.201402291
Abstract
Lysine methylation is one of the important post-translational modifications of histones, and produces an Nε-mono-, di-, or trimethyllysine residues. Multiple and site-specific lysine methylations of histones are essential to define epigenetic statuses and control heterochromatin formation, DNA repair, and transcription regulation. A method was previously developed to build an analogue of Nε-monomethyllysine, with cysteine substituting for lysine. Here, we have developed a new method of preparing histones bearing multiple Nε-monomethyllysine residues at specified positions. Release factor 1-knockout (RFzero) Escherichia coli cells or a cell-free system based on the RFzero cell lysate was used for protein synthesis, as in RFzero cells UAG is redefined as a sense codon for non-canonical amino acids. During protein synthesis, a tert-butyloxycarbonyl-protected Nε-monomethyllysine analogue is ligated to Methanosarcina mazei pyrrolysine tRNA (tRNAPyl) by M. mazei pyrrolysyl-tRNA synthetase mutants, and is translationally incorporated into one or more positions specified by the UAG codon. Protecting groups on the protein are then removed with trifluoroacetic acid to generate Nε-monomethyllysine residues. We installed Nε-monomethyllysine residues at positions 4, 9, 27, 36, and/or 79 of human histone H3. Each of the Nε-monomethyllysine residues within the produced histone H3 was recognized by its specific antibody. Furthermore, the antibody recognized the authentic Nε-monomethyllysine residue at position 27 better than the Nε-monomethyllysine analogue built with cysteine. Mass spectrometry analyses also confirmed the lysine modifications on the produced histone H3. Thus, our method enables the installation of authentic Nε-monomethyllysines at multiple positions within a protein for large-scale production.
Co-reporter:Tatsuo Yanagisawa;Takashi Umehara;Kensaku Sakamoto
ChemBioChem 2014 Volume 15( Issue 15) pp:2181-2187
Publication Date(Web):
DOI:10.1002/cbic.201402266
Co-reporter:Toshiaki Higo;Noriyuki Suka;Haruhiko Ehara
Journal of Structural and Functional Genomics 2014 Volume 15( Issue 4) pp:191-199
Publication Date(Web):2014 December
DOI:10.1007/s10969-014-9190-1
We developed a method for efficient chromosome tagging in Pichia pastoris, using a useful tandem affinity purification (TAP) tag. The TAP tag, designated and used here as the THF tag, contains a thrombin protease cleavage site for removal of the TAP tag and a hexahistidine sequence (6× His) followed by three copies of the FLAG sequence (3× FLAG) for affinity purification. Using this method, THF-tagged RNA polymerases I, II, and III were successfully purified from P. pastoris. The method also enabled us to purify the tagged RNA polymerase II on a large scale, for its crystallization and preliminary X-ray crystallographic analysis. The method described here will be widely useful for the rapid and large-scale preparation of crystallization grade eukaryotic multi-subunit protein complexes.
Co-reporter:Mitsuo Kuratani;Tatsuo Yanagisawa
Journal of Structural and Functional Genomics 2014 Volume 15( Issue 3) pp:173-180
Publication Date(Web):2014 September
DOI:10.1007/s10969-014-9183-0
The N1-methyladenosine residue at position 58 of tRNA is found in the three domains of life, and contributes to the stability of the three-dimensional L-shaped tRNA structure. In thermophilic bacteria, this modification is important for thermal adaptation, and is catalyzed by the tRNA m1A58 methyltransferase TrmI, using S-adenosyl-l-methionine (AdoMet) as the methyl donor. We present the 2.2 Å crystal structure of TrmI from the extremely thermophilic bacterium Aquifex aeolicus, in complex with AdoMet. There are four molecules per asymmetric unit, and they form a tetramer. Based on a comparison of the AdoMet binding mode of A. aeolicus TrmI to those of the Thermus thermophilus and Pyrococcus abyssi TrmIs, we discuss their similarities and differences. Although the binding modes to the N6 amino group of the adenine moiety of AdoMet are similar, using the side chains of acidic residues as well as hydrogen bonds, the positions of the amino acid residues involved in binding are diverse among the TrmIs from A. aeolicus, T. thermophilus, and P. abyssi.
Co-reporter:Hideaki Niwa;Junko Mikuni;Shunta Sasaki
Journal of Structural and Functional Genomics 2014 Volume 15( Issue 3) pp:153-164
Publication Date(Web):2014 September
DOI:10.1007/s10969-014-9188-8
Ribosomal protein S6 kinase 1 (S6K1) is a serine/threonine protein kinase that plays an important role in the PIK3/mTOR signaling pathway, and is implicated in diseases including diabetes, obesity, and cancer. The crystal structures of the S6K1 kinase domain in complexes with staurosporine and the S6K1-specific inhibitor PF-4708671 have been reported. In the present study, five compounds (F108, F109, F176, F177, and F179) were newly identified by in silico screening of a chemical library and kinase assay. The crystal structures of the five inhibitors in complexes with the S6K1 kinase domain were determined at resolutions between 1.85 and 2.10 Å. All of the inhibitors bound to the ATP binding site, lying along the P-loop, while the activation loop stayed in the inactive form. Compound F179, with a carbonyl group in the middle of the molecule, altered the αC helix conformation by interacting with the invariant Lys123. Compounds F176 and F177 bound slightly distant from the hinge region, and their sulfoamide groups formed polar interactions with the protein. The structural features required for the specific binding of inhibitors are discussed.
Co-reporter:Kazuhiro Kashiwagi;Takuhiro Ito
Journal of Structural and Functional Genomics 2014 Volume 15( Issue 3) pp:125-130
Publication Date(Web):2014 September
DOI:10.1007/s10969-014-9177-y
The eukaryotic translation initiation factor 2A (eIF2A) was identified as a factor that stimulates the binding of methionylated initiator tRNA (Met-tRNAiMet) to the 40S ribosomal subunit, but its physiological role remains poorly defined. Recently, eIF2A was shown to be involved in unconventional translation initiation from CUG codons and in viral protein synthesis under stress conditions where eIF2 is inactivated. We determined the crystal structure of the WD-repeat domain of Schizosaccharomyces pombe eIF2A at 2.5 Å resolution. The structure adopts a novel nine-bladed β-propeller fold. In contrast to the usual β-propeller proteins, the central channel of the molecule has the narrower opening on the bottom of the protein and the wider opening on the top. Highly conserved residues are concentrated in the positively-charged top face, suggesting the importance of this face for interactions with nucleic acids or other initiation factors.
Co-reporter:Markus J. Bröcker;Yuzuru Itoh;Shun-ichi Sekine;Gifty Hammond;Shiro Suetsugu;Dieter Söll
Science 2013 Volume 340(Issue 6128) pp:75-78
Publication Date(Web):05 Apr 2013
DOI:10.1126/science.1229521
Putting Selenium in Proteins
The 21st amino acid, selenocysteine (Sec), occurs in the active site of many redox enzymes. Its cognate transfer RNA (tRNA) is first loaded with Ser by seryl-tRNA synthetase and the Ser-tRNASec is then converted to Sec-tRNASec. Itoh et al. (p. 75) determined the crystal structures of the selenocysteine synthase, SelA, that is responsible for this conversion in bacteria, alone and in complex with tRNA. The decameric SelA complex binds to 10 tRNASec molecules. The structures, together with biochemistry, show how SelA discriminates tRNASec from tRNASer, give insight into the mechanism of catalysis, and show that decamerization is essential for function.
Co-reporter:Shun-ichi Sekine, Yuko Murayama, Vladimir Svetlov, Evgeny Nudler, Shigeyuki Yokoyama
Molecular Cell (5 February 2015) Volume 57(Issue 3) pp:408-421
Publication Date(Web):5 February 2015
DOI:10.1016/j.molcel.2014.12.014
•RNA polymerase can assume the tight and ratcheted forms (T and R)•The R form is for GreA-dependent RNA cleavage, arrest, and hairpin-pause/termination•The T form is for GreA-independent RNA cleavage, nucleotide addition, and its reversal•The paused/backtracked T form readily transitions to the R form, governed by RNADNA-dependent RNA polymerase (RNAP) accomplishes multiple tasks during transcription by assuming different structural forms. Reportedly, the “tight” form performs nucleotide addition to nascent RNA, while the “ratcheted” form is adopted for transcription inhibition. In this study, we performed Cys-pair crosslinking (CPX) analyses of various transcription complexes of a bacterial RNAP and crystallographic analyses of its backtracked and Gre-factor-bound states to clarify which of the two forms is adopted. The ratcheted form was revealed to support GreA-dependent transcript cleavage, long backtracking, hairpin-dependent pausing, and termination. In contrast, the tight form correlated with nucleotide addition, mismatch-dependent pausing, one-nucleotide backtracking, and factor-independent transcript cleavage. RNAP in the paused/backtracked state, but not the nucleotide-addition state, readily transitions to the ratcheted form (“ratchetable”), indicating that the tight form represents two distinct regulatory states. The 3′ end and the hairpin structure of the nascent RNA promote the ratchetable nature by modulating the trigger-loop conformation.Download high-res image (614KB)Download full-size image
Co-reporter:Yasushi Hikida, Michiko Kimoto, Ichiro Hirao, Shigeyuki Yokoyama
Biochemical and Biophysical Research Communications (29 January 2017) Volume 483(Issue 1) pp:52-57
Publication Date(Web):29 January 2017
DOI:10.1016/j.bbrc.2017.01.007
Co-reporter:Seisuke Kusano, Mutsuko Kukimoto-Niino, Yoko Satta, Noboru Ohsawa, ... Shigeyuki Yokoyama
Journal of Molecular Biology (26 August 2014) Volume 426(Issue 17) pp:3016-3027
Publication Date(Web):26 August 2014
DOI:10.1016/j.jmb.2014.06.020
•Structural basis for non-self-antigen recognition by class II HLA-DP was unknown.•Crystal structure of HLA-DP5 complexed with the pollen allergen Cry j 1 was solved.•The mechanisms of the recognition of the nine-residue allergen peptide were elucidated.•In particular, the acidic P1 pocket of HLA-DP5 fully accommodates the Lys side chain.•The HLA-DP family is divided into the DP5 and DP2 groups, based on recognition modes.The major allergen, Cry j 1, was isolated from Japanese cedar Cryptomeria japonica (Cry j) pollen and was shown to react with immunoglobulin E antibodies in the sera from pollinosis patients. We previously reported that the frequency of HLA-DP5 was significantly higher in pollinosis patients and the immunodominant peptides from Cry j 1 bound to HLA-DP5 to activate Th2 cells. In the present study, we determined the crystal structure of the HLA-DP5 heterodimer in complex with a Cry j 1-derived nine-residue peptide, at 2.4 Å resolution. The peptide-binding groove recognizes the minimal peptide with 10 hydrogen bonds, including those between the negatively charged P1 pocket and the Lys side chain at the first position in the peptide sequence. We confirmed that HLA-DP5 exhibits the same Cry j 1-binding mode in solution, through pull-down experiments using structure-based mutations of Cry j 1. We also identified the characteristic residues of HLA-DP5 that are responsible for the distinct properties of the groove, by comparing the structure of HLA-DP5 and the previously reported structures of HLA-DP2 in complexes with pDRA of the self-antigen. The comparison revealed that the HLA-DP5·pCry j 1 complex forms several hydrogen bond/salt bridge networks between the receptor and the antigen that were not observed in the HLA-DP2·pDRA complex. Evolutionary considerations have led us to conclude that HLA-DP5 and HLA-DP2 represent two major groups of the HLA-DP family, in which the properties of the P1 and P4 pockets have evolved and acquired the present ranges of epitope peptide-binding specificities.Download high-res image (165KB)Download full-size image
Co-reporter:Shisako Shoji, Yutaka Muto, Mariko Ikeda, Fahu He, ... Shigeyuki Yokoyama
FEBS Open Bio (2014) Volume 4() pp:689-703
Publication Date(Web):1 January 2014
DOI:10.1016/j.fob.2014.06.010
•Overexpression of the ZBR fragment of Emi2, but not of Emi1, induces abnormal cell division.•The Emi2 ZBR fragment impairs the association of the coactivator Cdc20 with APC/C.•The Emi2 ZBR fragment inhibits ubiquitylation by the cullin-RING of APC/C and E2C.•The Emi2 ZBR-specific residues for APC/C inhibitory activity have been identified.Anaphase-promoting complex or cyclosome (APC/C) is a multisubunit ubiquitin ligase E3 that targets cell-cycle regulators. Cdc20 is required for full activation of APC/C in M phase, and mediates substrate recognition. In vertebrates, Emi2/Erp1/FBXO43 inhibits APC/C-Cdc20, and functions as a cytostatic factor that causes long-term M phase arrest of mature oocytes. In this study, we found that a fragment corresponding to the zinc-binding region (ZBR) domain of Emi2 inhibits cell-cycle progression, and impairs the association of Cdc20 with the APC/C core complex in HEK293T cells. Furthermore, we revealed that the ZBR fragment of Emi2 inhibits in vitro ubiquitin chain elongation catalyzed by the APC/C cullin-RING ligase module, the ANAPC2–ANAPC11 subcomplex, in combination with the ubiquitin chain-initiating E2, E2C/UBE2C/UbcH10. Structural analyses revealed that the Emi2 ZBR domain uses different faces for the two mechanisms. Thus, the double-faced ZBR domain of Emi2 antagonizes the APC/C function by inhibiting both the binding with the coactivator Cdc20 and ubiquitylation mediated by the cullin-RING ligase module and E2C. In addition, the tail region between the ZBR domain and the C-terminal RL residues [the post-ZBR (PZ) region] interacts with the cullin subunit, ANAPC2. In the case of the ZBR fragment of the somatic paralogue of Emi2, Emi1/FBXO5, these inhibitory activities against cell division and ubiquitylation were not observed. Finally, we identified two sets of key residues in the Emi2 ZBR domain that selectively exert each of the dual Emi2-specific modes of APC/C inhibition, by their mutation in the Emi2 ZBR domain and their transplantation into the Emi1 ZBR domain.
Co-reporter:Fahu He, Kengo Tsuda, Mari Takahashi, Kanako Kuwasako, ... Yutaka Muto
FEBS Letters (2 November 2012) Volume 586(Issue 21) pp:3858-3864
Publication Date(Web):2 November 2012
DOI:10.1016/j.febslet.2012.09.009
The WWE domain is often identified in proteins associated with ubiquitination or poly-ADP-ribosylation. Structural information about WWE domains has been obtained for the ubiquitination-related proteins, such as Deltex and RNF146, but not yet for the poly-ADP-ribose polymerases (PARPs). Here we determined the solution structures of the WWE domains from PARP11 and PARP14, and compared them with that of the RNF146 WWE domain. NMR perturbation experiments revealed the specific differences in their ADP-ribose recognition modes that correlated with their individual biological activities. The present structural information sheds light on the ADP-ribose recognition modes by the PARP WWE domains.Highlights► First reported solution structures of WWE domains from two different functional subfamilies. ► The solution structure of the WWE domain with ATP reveals the specific adenine recognition mode. ► NMR experiments reveal the binding preference for ADP-ribose among the WWE subfamily members.
Co-reporter:Yuzuru Itoh, Markus J. Bröcker, Shun-ichi Sekine, Dieter Söll, Shigeyuki Yokoyama
Journal of Molecular Biology (17 April 2014) Volume 426(Issue 8) pp:1723-1735
Publication Date(Web):17 April 2014
DOI:10.1016/j.jmb.2014.01.003
•The catalytic sites of SelA are close to the dimer pentamerization interfaces.•Mutations in the dimer–dimer interface resulted in loss of SelA activity.•The crystal structures of “depentamerized” dimeric SelA mutants were determined.•Dimeric SelA exhibits a deformed and inactivated active site.•The decameric quaternary structure was evolutionally acquired for the SelA activity.The 21st amino acid, selenocysteine (Sec), is incorporated translationally into proteins and is synthesized on its specific tRNA (tRNASec). In Bacteria, the selenocysteine synthase SelA converts Ser-tRNASec, formed by seryl-tRNA synthetase, to Sec-tRNASec. SelA, a member of the fold-type-I pyridoxal 5′-phosphate-dependent enzyme superfamily, has an exceptional homodecameric quaternary structure with a molecular mass of about 500 kDa. Our previously determined crystal structures of Aquifex aeolicus SelA complexed with tRNASec revealed that the ring-shaped decamer is composed of pentamerized SelA dimers, with two SelA dimers arranged to collaboratively interact with one Ser-tRNASec. The SelA catalytic site is close to the dimer–dimer interface, but the significance of the dimer pentamerization in the catalytic site formation remained elusive. In the present study, we examined the quaternary interactions and demonstrated their importance for SelA activity by systematic mutagenesis. Furthermore, we determined the crystal structures of “depentamerized” SelA variants with mutations at the dimer–dimer interface that prevent pentamerization. These dimeric SelA variants formed a distorted and inactivated catalytic site and confirmed that the pentamer interactions are essential for productive catalytic site formation. Intriguingly, the conformation of the non-functional active site of dimeric SelA shares structural features with other fold-type-I pyridoxal 5′-phosphate-dependent enzymes with native dimer or tetramer (dimer-of-dimers) quaternary structures.Download high-res image (143KB)Download full-size image

![L-Lysine, N6-[[(3-azido-5-nitrophenyl)methoxy]carbonyl]-N2-[(9H-fluoren-9-ylmethoxy)carbonyl]-](/data/chemimg/3723200/1836202-32-4.png)
![L-Lysine, N6-[[(3-azido-5-nitrophenyl)methoxy]carbonyl]-N2-[(9H-fluoren-9-ylmethoxy)carbonyl]-](/data/chemimg/3723200/1836202-32-4_b.png)
![L-Lysine, N6-[[[3-azido-5-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]phenyl]methoxy]carbonyl]-N2-[(9H-fluoren-9-ylmethoxy)carbonyl]-](/data/chemimg/3723200/1836202-30-2.png)
![L-Lysine, N6-[[[3-azido-5-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]phenyl]methoxy]carbonyl]-N2-[(9H-fluoren-9-ylmethoxy)carbonyl]-](/data/chemimg/3723200/1836202-30-2_b.png)
![L-Lysine, N6-[[(3-azidophenyl)methoxy]carbonyl]-N2-[(9H-fluoren-9-ylmethoxy)carbonyl]-](/data/chemimg/3723200/1836202-27-7.png)
![L-Lysine, N6-[[(3-azidophenyl)methoxy]carbonyl]-N2-[(9H-fluoren-9-ylmethoxy)carbonyl]-](/data/chemimg/3723200/1836202-27-7_b.png)
![L-Lysine, N6-[[(3-azido-5-nitrophenyl)methoxy]carbonyl]-](/data/chemimg/3723200/1836202-33-5.png)
![L-Lysine, N6-[[(3-azido-5-nitrophenyl)methoxy]carbonyl]-](/data/chemimg/3723200/1836202-33-5_b.png)
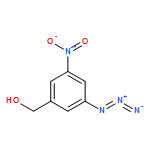
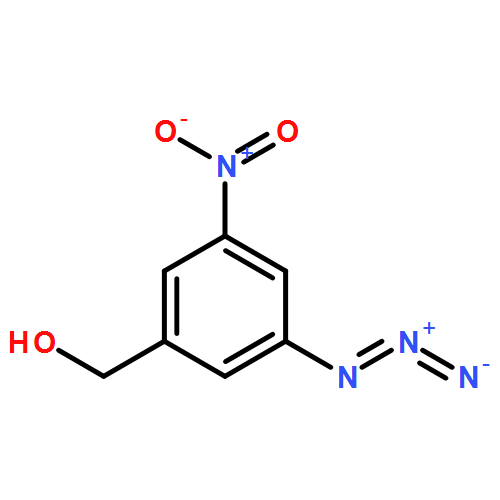
![Carbamic acid, N-[3-azido-5-(hydroxymethyl)phenyl]-, 9H-fluoren-9-ylmethyl ester](/data/chemimg/3723200/1836202-29-9.png)
![Carbamic acid, N-[3-azido-5-(hydroxymethyl)phenyl]-, 9H-fluoren-9-ylmethyl ester](/data/chemimg/3723200/1836202-29-9_b.png)
![L-Lysine, N6-[[(3-azidophenyl)methoxy]carbonyl]-](/data/chemimg/3723200/1836202-28-8.png)
![L-Lysine, N6-[[(3-azidophenyl)methoxy]carbonyl]-](/data/chemimg/3723200/1836202-28-8_b.png)
![L-Lysine, N6-[[(3-amino-5-azidophenyl)methoxy]carbonyl]-](/data/chemimg/3723200/1836202-31-3.png)
![L-Lysine, N6-[[(3-amino-5-azidophenyl)methoxy]carbonyl]-](/data/chemimg/3723200/1836202-31-3_b.png)
![Piperidine, 1-acetyl-4-(5H-dibenzo[a,d]cyclohepten-5-ylidene)-](http://img.cochemist.com/ccimg/117600/117540-06-4.png)
![Piperidine, 1-acetyl-4-(5H-dibenzo[a,d]cyclohepten-5-ylidene)-](http://img.cochemist.com/ccimg/117600/117540-06-4_b.png)
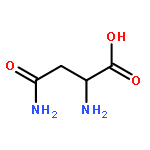

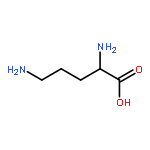
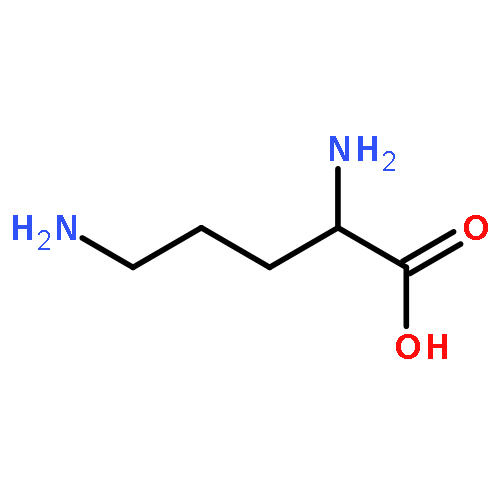
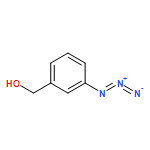
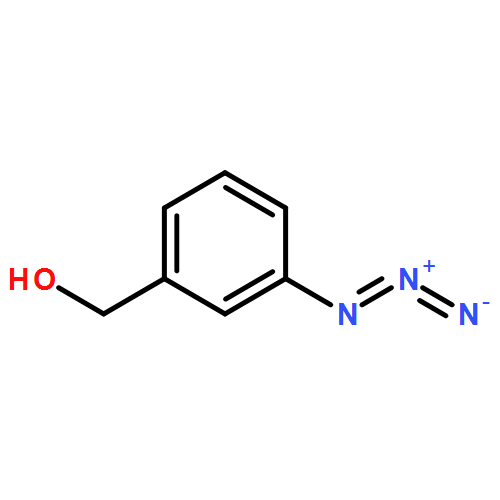
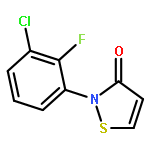




![N-[3-(4-nitrophenyl)-3-oxoprop-1-en-2-yl]acetamide](http://img.cochemist.com/ccimg/6700/6627-80-1.png)
![N-[3-(4-nitrophenyl)-3-oxoprop-1-en-2-yl]acetamide](http://img.cochemist.com/ccimg/6700/6627-80-1_b.png)


![7H-Pyrrolo[2,3-d]pyrimidin-4-amine,7-cyclopentyl-5-(4-phenoxyphenyl)-](http://img.cochemist.com/ccimg/213800/213743-31-8.png)
![7H-Pyrrolo[2,3-d]pyrimidin-4-amine,7-cyclopentyl-5-(4-phenoxyphenyl)-](http://img.cochemist.com/ccimg/213800/213743-31-8_b.png)

![Imidazo[2,1-b]thiazole,6-(4-fluorophenyl)-2,3-dihydro-5-(4-pyridinyl)-, hydrochloride (1:2)](http://img.cochemist.com/ccimg/116400/116339-68-5.png)
![Imidazo[2,1-b]thiazole,6-(4-fluorophenyl)-2,3-dihydro-5-(4-pyridinyl)-, hydrochloride (1:2)](http://img.cochemist.com/ccimg/116400/116339-68-5_b.png)
![Benzene, 1-[(4-chlorophenyl)sulfonyl]-2,4-dinitro-](http://img.cochemist.com/ccimg/900/899-03-6.png)
![Benzene, 1-[(4-chlorophenyl)sulfonyl]-2,4-dinitro-](http://img.cochemist.com/ccimg/900/899-03-6_b.png)
![4-(5H-Dibenzo[a,d][7]annulen-5-ylidene)-1-methylpiperidine](http://img.cochemist.com/ccimg/200/129-03-3.png)
![4-(5H-Dibenzo[a,d][7]annulen-5-ylidene)-1-methylpiperidine](http://img.cochemist.com/ccimg/200/129-03-3_b.png)














![3H-Imidazo[4,5-b]pyridine,7-(2-thienyl)-](http://img.cochemist.com/ccimg/901200/901130-24-3.png)
![3H-Imidazo[4,5-b]pyridine,7-(2-thienyl)-](http://img.cochemist.com/ccimg/901200/901130-24-3_b.png)