Co-reporter:Sen-Ichi Aizawa;Soshi Tsubosaka
Chirality 2016 Volume 28( Issue 1) pp:85-91
Publication Date(Web):
DOI:10.1002/chir.22546
Abstract
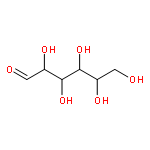
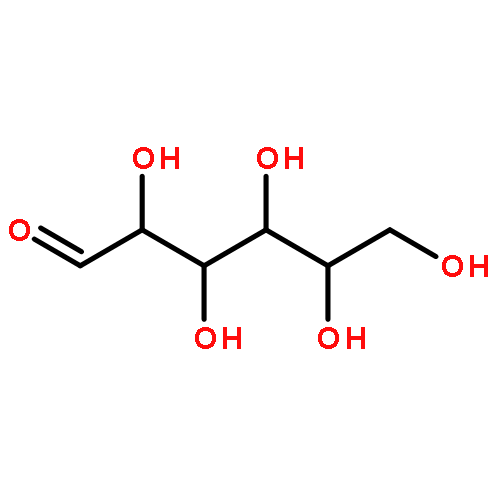
The optically active mixed-ligand fac(S)-tris(thiolato)rhodium(III) complexes, ΔL-fac(S)-[Rh(aet)2(L-cys-N,S)]− (aet = 2-aminoethanethiolate, L-cys = L-cysteinate) (1) and ΔLL-fac(S)-[Rh(aet)(L-cys-N,S)2]2− were newly prepared by the equatorial preference of the carboxyl group in the coordinated L-cys ligand. The amide formation reaction of 1 with 1,10-diaminodecane and polyallylamine gave the diamine-bridged dinuclear Rh(III) complex and the single-chain polymer-supported Rh(III) complex with retention of the ΔL configuration of 1, respectively. These Rh(III) complexes reacted with Co(III) or Co(II) to give the linear-type trinuclear structure with the S-bridged Co(III) center and the two Δ-Rh(III) terminal moieties. The polymer-supported Rh(III) complex was applied not only to the CD spectropolarimetric detection and determination of a trace of precious metal ions such as Au(III), Pt(II), and Pd(II) but also to concentration and extraction of these metal ions into the solid polymer phase. Chirality 28:85–91, 2016. © 2015 Wiley Periodicals, Inc.
Co-reporter:Sen-Ichi Aizawa;Takahiro Kidani;Sayuri Takada ;Yumika Ofusa
Chirality 2015 Volume 27( Issue 5) pp:353-357
Publication Date(Web):
DOI:10.1002/chir.22443
Abstract
Readily available L-tartaric acid, which is a bidentate ligand with two chiral centers forming a seven-membered chelate ring, was applied to the chiral ligand for the chiral nuclear magnetic resonance (NMR) shift reagent of samarium(III) formed in situ. This simple method does not cause serious signal broadening in the high magnetic field. Enantiomeric 13C and 1H NMR signals and enantiotopic 1H NMR signals of α-amino acids were successfully resolved at pH 8.0 and the 1:3 molar ratio of Sm(NO3)3:L-tartaric acid. It is elucidated that the enantiomeric signal resolution is attributed to the anisotropic magnetic environment for the enantiomers induced by the chiral L-tartarato samarium(III) complex rather than differences in stability of the diastereomeric substrate adducts. The present 13C NMR signal resolution was also effective for the practical simultaneous analysis of plural kinds of DL-amino acids. Chirality 27:353–357, 2015.© 2015 Wiley Periodicals, Inc.
Co-reporter:Sen-ichi Aizawa, Koichi Fukumoto, Tatsuya Kawamoto
Polyhedron 2013 Volume 62() pp:37-41
Publication Date(Web):7 October 2013
DOI:10.1016/j.poly.2013.06.013
Tris[2-(diphenylphosphino)ethyl]phosphine disulfide (pp3S2), in which two terminal phosphino groups are selectively sulfidated, was prepared by utilizing the selective sulfidation reaction of [PdI(pp3)]I. Co(II) complexes with bidentate, tridentate and tetradentate phosphines and pp3S2 were prepared from anhydrous CoI2. X-ray crystal analyses revealed that the reaction of CoI2 with 1 equivalent of 1,2-bis(diphenylphosphino)ethane (p2) gave rise to partial oxidation of p2 to give the dicationic octahedral [Co(p2O2)2(CH3CN)2]2+ (p2O2 = p2 dioxide) and dianionic p2O-bridged tetrahedral dinuclear [CoI3(p2O)CoI3]2− (p2O = p2 monooxide) complexes, while the reaction with 2 equivalents of p2 gave the square-pyramidal [CoI(p2)2]+ complex. The catalytic activity for the Co-catalyzed coupling reaction of 2-iodobutane with n-butyl acrylate was compared using multidentate phosphines and phosphine sulfides as ligands, and the efficiency of the phosphine sulfides was shown. The tendency for multidentate phosphine to deactivate the Co-catalysis can substantiate an oxidative addition driven mechanism in which the multidentate ligand should interfere with the formation of the alkyl halide Co(III) adduct and subsequent coordination of an alkene.Steric and electronic effects of phosphine and phosphine sulfide ligands on the cobalt-catalyzed reductive coupling of an alkyl halide, 2-iodobutane, with an activated alkene, n-butyl acrylate, are reported. The observed differences in the catalytic activity provide insight into the reaction mechanism of the catalytic cycle and knowledge for choosing the ligand of the cobalt catalyst.
Co-reporter:Sen-ichi Aizawa, Tatsuya Kawamoto, Suji Nishigaki, Ayano Sasaki
Journal of Organometallic Chemistry 2011 696(11–12) pp: 2471-2476
Publication Date(Web):
DOI:10.1016/j.jorganchem.2011.03.018
Co-reporter:Sen-ichi Aizawa, Tatsuya Kawamoto, Yuuto Asai, Chie Ishimura
Journal of Organometallic Chemistry 2010 695(8) pp: 1253-1260
Publication Date(Web):
DOI:10.1016/j.jorganchem.2010.02.006
Co-reporter:Sen-ichi Aizawa, Arpi Majumder, Yukihiro Yokoyama, Mitsuyoshi Tamai, Daisuke Maeda and Akina Kitamura
Organometallics 2009 Volume 28(Issue 20) pp:6067-6072
Publication Date(Web):September 25, 2009
DOI:10.1021/om900588v
Mononuclear phosphine sulfide Pd(0) complexes and a polymer-supported triphenylphosphine sulfide Pd(0) complex were prepared as new air-stable Pd(0) catalysts for C−C coupling reactions. The phosphine sulfide Pd(0) complexes are not decomposed after completion of Suzuki−Miyaura coupling, and the polymer-supported Pd(0) catalyst is practically recyclable, while phosphine Pd(0) complexes are decomposed into inactive Pd(0) black after consuming the substrates. New catalytic activity of Pd(0) that promotes chalcogen atom replacement of phosphine chalcogenides (R3P═X, X = O, S, Se) is reported. A mechanistic study revealed that the new catalytic chalcogen replacement results from activation of the P═X bond as well as promotion of the oxidative chalcogenide formation. The intermediate phosphine was successfully trapped as a phosphine Pd(II) complex, and the P═X bond activation is applicable to regeneration of phosphine or phosphine sulfide from oxidized phosphine.
Co-reporter:Sen-ichi Aizawa, Mayumi Kondo, Ryuta Miyatake, Mitsuyoshi Tamai
Inorganica Chimica Acta 2007 Volume 360(Issue 8) pp:2809-2813
Publication Date(Web):30 May 2007
DOI:10.1016/j.ica.2006.12.046
A phosphine sulfide Pd(II) complex, [Pd(p2S2)2](BF4)2 (1) (p2S2 = 1,2-bis(diphenylphosphino)ethane disulfide), was synthesized and characterized by an X-ray crystal structure analysis and 31P NMR spectroscopy. The p2S2 ligand exchange rate of 1 with free p2S2 in chloroform was revealed to be comparable to the general solvent exchange rate on Pd(II). The catalytic activity of 1 was evaluated by carrying out the Heck reaction. The diminishing of the induction period and acceleration of the reaction were observed for 1 by comparing the phosphine Pd(II) complexes with a leaving chloro ligand, [PdCl(p3)]Cl (p3 = bis[2-(diphenylphosphino)ethyl]phenylphosphine) and [PdCl(pp3)]Cl (pp3 = tris[2-(diphenylphosphino)ethyl]phosphine), and the catalytic activity was comparable to that of the phosphine Pd(0) complex, [Pd(PPh3)4]. Such a high catalytic activity of 1 is attributed to the π-accepting ability of the phosphine sulfide S atom which stabilizes the catalytically active Pd(0) species electronically and weak σ-donation of the S atom which does not block the formation and a subsequent reaction of the Pd(II) substrate adduct in the catalytic cycle.A novel phosphine sulfide Pd(II) complex with 1,2-bis(diphenylphosphino)ethane disulfide was employed as a catalyst for the C–C coupling reaction. π accepting ability for the phosphine sulfide group promotes the prereduction of the Pd(II) and their weak σ donation does not block the catalytic cycle on the Pd(II) substrate adduct.