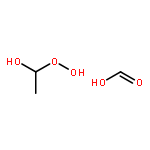
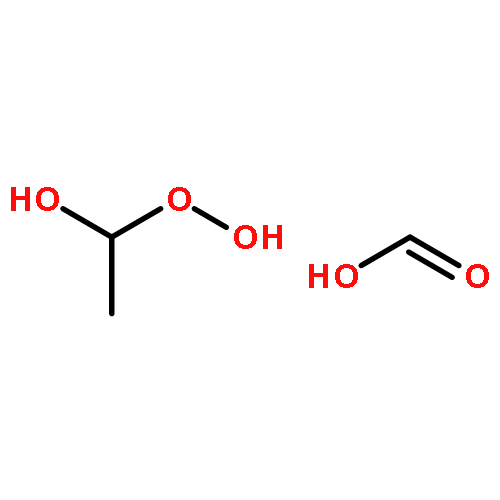
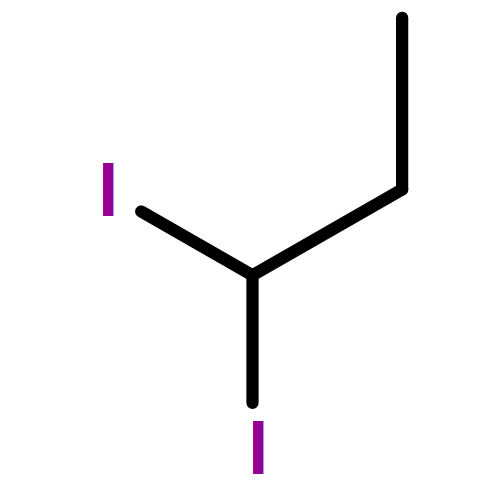
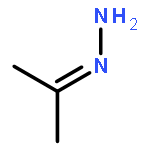
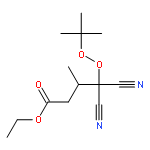
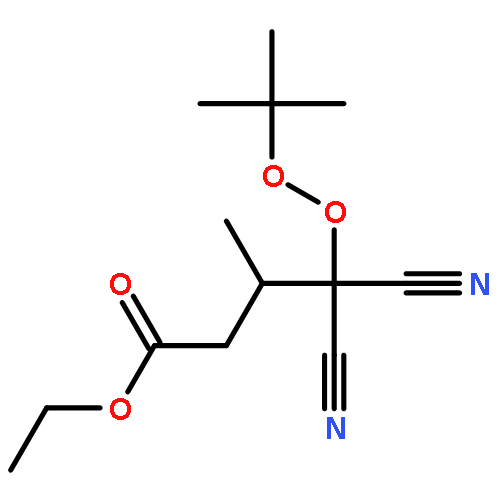
Co-reporter:Greg T. Drozd, Theo Kurtén, Neil M. Donahue, and Marsha I. Lester
The Journal of Physical Chemistry A August 17, 2017 Volume 121(Issue 32) pp:6036-6036
Publication Date(Web):July 10, 2017
DOI:10.1021/acs.jpca.7b05495
We used the steady-state master equation to model unimolecular decay of the Criegee intermediate formed from ozonolysis of 2,3-dimethyl-2-butene (tetramethylethylene, TME). Our results show the relative importance and time scales for both the prompt and thermal unimolecular decay of the dimethyl-substituted Criegee intermediate, (CH3)2COO. Calculated reactive fluxes show the importance of quantum mechanical tunneling for both prompt and thermal decay to OH radical products. We constrained the initial energy distribution of chemically activated (CH3)2COO formed in TME ozonolysis by combining microcanonical rates k(E) measured experimentally under collision-free conditions and modeled using semiclassical transition-state theory (SCTST) with pressure-dependent yields of stabilized Criegee intermediates measured with scavengers in flow-tube experiments. Thermal decay rates under atmospheric conditions k(298 K, 1 atm) increase by more than 1 order of magnitude when tunneling is included. Accounting for tunneling has important consequences for interpreting pressure dependent yields of stabilized Criegee intermediates, particularly with regard to the fraction of Criegee intermediates formed in the zero-pressure limit.
Co-reporter:Craig A. Taatjes, Fang Liu, Brandon Rotavera, Manoj Kumar, Rebecca Caravan, David L. Osborn, Ward H. Thompson, and Marsha I. Lester
The Journal of Physical Chemistry A 2017 Volume 121(Issue 1) pp:
Publication Date(Web):December 21, 2016
DOI:10.1021/acs.jpca.6b07712
Hydroxyacetone (CH3C(O)CH2OH) is observed as a stable end product from reactions of the (CH3)2COO Criegee intermediate, acetone oxide, in a flow tube coupled with multiplexed photoionization mass spectrometer detection. In the experiment, the isomers at m/z = 74 are distinguished by their different photoionization spectra and reaction times. Hydroxyacetone is observed as a persistent signal at longer reaction times at a higher photoionization threshold of ca. 9.7 eV than Criegee intermediate and definitively identified by comparison with the known photoionization spectrum. Complementary electronic structure calculations reveal multiple possible reaction pathways for hydroxyacetone formation, including unimolecular isomerization via hydrogen atom transfer and −OH group migration as well as self-reaction of Criegee intermediates. Varying the concentration of Criegee intermediates suggests contributions from both unimolecular and self-reaction pathways to hydroxyacetone. The hydroxyacetone end product can provide an effective, stable marker for the production of transient Criegee intermediates in future studies of alkene ozonolysis.
Co-reporter:Fang Liu, Yi Fang, Manoj Kumar, Ward H. Thompson and Marsha I. Lester
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 32) pp:20490-20494
Publication Date(Web):22 Jul 2015
DOI:10.1039/C5CP02917A
Many alkyl-substituted Criegee intermediates are predicted to undergo an intramolecular 1,4-hydrogen transfer to form isomeric vinyl hydroperoxide species (CCOOH moiety), which break apart to release OH and vinoxy radicals. We report direct detection of stabilized vinyl hydroperoxides formed via carboxylic acid-catalyzed tautomerization of Criegee intermediates. A doubly hydrogen-bonded interaction between the Criegee intermediate and carboxylic acid facilitates efficient hydrogen transfer through a double hydrogen shift. Deuteration of formic or acetic acid permits migration of a D atom to yield partially deuterated vinyl hydroperoxides, which are distinguished from the CH3CHOO, (CH3)2COO, and CH3CH2CHOO Criegee intermediates by mass. Using 10.5 eV photoionization, three prototypical vinyl hydroperoxides, CH2CHOOD, CH2C(CH3)OOD, and CH3CHCHOOD, are detected directly. Complementary electronic structure calculations reveal several reaction pathways, including the barrierless acid-catalyzed tautomerization reaction predicted previously and a barrierless addition reaction that yields hydroperoxy alkyl formate.
Co-reporter:Hongwei Li, Yi Fang, Nathanael M. Kidwell, Joseph M. Beames, and Marsha I. Lester
The Journal of Physical Chemistry A 2015 Volume 119(Issue 30) pp:8328-8337
Publication Date(Web):July 20, 2015
DOI:10.1021/acs.jpca.5b05352
UV excitation of jet-cooled CH3CHOO on the B1A′–X1A′ transition results in dissociation to two spin-allowed product channels: CH3CHO X1A′ + O 1D and CH3CHO a3A″ + O 3P. The O 1D and O 3P products are detected using 2 + 1 REMPI at 205 and 226 nm, respectively, for action spectroscopy and velocity map imaging studies. The O 1D action spectrum closely follows the previously reported UV absorption spectrum for jet-cooled CH3CHOO [Beames et al. J. Chem. Phys. 2013, 138, 244307]. Velocity map images of the O 1D products following excitation of CH3CHOO at 305, 320, and 350 nm exhibit anisotropic angular distributions indicative of rapid (ps) dissociation, along with broad and unstructured total kinetic energy (TKER) distributions that reflect the internal energy distribution of the CH3CHO X1A′ coproducts. The O 3P action spectrum turns on near the peak of the UV absorption spectrum (ca. 324 nm) and extends to higher energy with steadily increasing O 3P yield. Excitation of CH3CHOO at 305 nm, attributed to absorption of the more stable syn-conformer, also results in an anisotropic angular distribution of O 3P products arising from rapid (ps) dissociation, but a narrower TKER distribution since less energy is available to the CH3CHO a3A″ + O 3P products. The threshold for the higher energy CH3CHO a3A″ + O 3P product channel is determined to be ca. 88.4 kcal mol–1 from the termination of the TKER distribution and the onset of the O 3P action spectrum. This threshold is combined with the singlet-triplet spacings of O atoms and acetaldehyde to establish the dissociation energy for syn-CH3HOO X1A′ to the lowest spin-allowed product channel, CH3CHO X1A′ + O 1D, of ≤55.9 ± 0.4 kcal mol–1. A harmonic normal-mode analysis is utilized to identify the vibrational modes of CH3CHO likely to be excited upon dissociation into the two product channels.
Co-reporter:Lu Lu, Joseph M. Beames, Marsha I. Lester
Chemical Physics Letters 2014 Volume 598() pp:23-27
Publication Date(Web):8 April 2014
DOI:10.1016/j.cplett.2014.02.049
•Early time detection of OH products from unimolecular decay of Criegee intermediates.•Doppler broadened line profiles for OH products from CH3CHOO intermediates.•Predominant translational excitation of products from CH3CHOO intermediates.•Substantially different decay mechanisms for energized CH2OO and CH3CHOO.The OH radical products from unimolecular decay of energized CH2OO and CH3CHOO Criegee intermediates are detected by laser-induced fluorescence shortly after they are formed utilizing 248 nm photolysis of diiodo precursors and subsequent reaction with O2. The OH radicals from both CH2OO and CH3CHOO are produced with average rotational energies of ∼1–2 kcal mol−1. A different mechanism for unimolecular decay of CH3CHOO is evident from noticeably broader OH A-X (1,0) line profiles, which is attributed to Doppler broadening. Modeling of the Doppler profiles suggests that most of the available energy is channeled into translational excitation of the products.
Co-reporter:Fang Liu;Joseph M. Beames;Andrew S. Petit;Anne B. McCoy
Science 2014 Vol 345(6204) pp:1596-1598
Publication Date(Web):26 Sep 2014
DOI:10.1126/science.1257158
Breaking down a Criegee intermediate
Ozone's damaging role in the upper atmosphere is well known, but ozone is also quite active closer down to where we live. In particular, ozone's run-ins with airborne unsaturated hydrocarbons, from natural or anthropogenic sources, produce even more-reactive OH radicals. Liu et al. used vibrational spectroscopy to study how OH emerges from a so-called Criegee intermediate formed when ozone attacks 2-butene. The results suggest that OH production is easier than current theory predicts.
Science, this issue p. 1596
Co-reporter:Fang Liu, Joseph M. Beames, Amy M. Green, and Marsha I. Lester
The Journal of Physical Chemistry A 2014 Volume 118(Issue 12) pp:2298-2306
Publication Date(Web):March 13, 2014
DOI:10.1021/jp412726z
Dimethyl- and ethyl-substituted Criegee intermediates, (CH3)2COO and CH3CH2CHOO, are photolytically generated from diiodo precursors, detected by VUV photoionization at 118 nm, and spectroscopically characterized via UV-induced depletion of the m/z = 74 signals under jet-cooled conditions. In each case, UV excitation resonant with the B–X transition results in significant ground-state depletion, reflecting the large absorption cross section and rapid dynamics in the excited B state. The broad UV absorption spectra of both (CH3)2COO and CH3CH2CHOO peak at ∼320 nm with absorption cross sections approaching ∼4 × 10–17 cm2 molec–1. The UV absorption spectra for (CH3)2COO and CH3CH2CHOO are similar to that reported previously for syn-CH3CHOO, suggesting analogous intramolecular interactions between the α-H and terminal O of the COO groups. Hydroxyl radical products generated concurrently with the Criegee intermediates are detected by 1 + 1′ resonance enhanced multiphoton ionization. The OH signals, scaled relative to those for the Criegee intermediates, are compared with prior studies of OH yield from alkene ozonolysis. The stationary points along the reaction coordinates from the alkyl-substituted Criegee intermediates to vinyl hydroperoxides and OH products are also computed to provide insight on the OH yields.
Co-reporter:Julia H. Lehman, Hongwei Li, Marsha I. Lester
Chemical Physics Letters 2013 590() pp: 16-21
Publication Date(Web):
DOI:10.1016/j.cplett.2013.10.029
Co-reporter:Julia H. Lehman, Marsha I. Lester, Jacek Kłos, Millard H. Alexander, Paul J. Dagdigian, Diego Herráez-Aguilar, F. Javier Aoiz, Mark Brouard, Helen Chadwick, Tom Perkins, and Scott A. Seamons
The Journal of Physical Chemistry A 2013 Volume 117(Issue 50) pp:13481-13490
Publication Date(Web):August 21, 2013
DOI:10.1021/jp407035p
Electronic quenching of OH A 2Σ+ by Kr was investigated through experimental studies of the collision cross sections and the OH X 2Π product state distribution. The quenching cross sections decrease with increasing rotational excitation in the excited OH A 2Σ+ electronic state. The OH X 2Π products of quenching exhibit a significant degree of rotational excitation but minimal vibrational excitation. Complementary theoretical studies of the OH (A 2Σ+, X 2Π) + Kr potential energy surfaces (PESs), nonadiabatic coupling, and quasiclassical trajectory calculations were carried out to elucidate the quenching dynamics. Accurate PESs for the two lowest diabatic states of A′ symmetry were computed along with the angularly dependent coupling between them. Coupling in nearly linear HO–Kr configurations provides the mechanism for the observed electronic quenching. A deep attractive well on the OH A 2Σ+ + Kr PES facilitates access to this region of strong coupling. Surface-hopping quasiclassical trajectory calculations yielded quenching cross sections and a OH X 2Π product rotational distribution in good accord with experimental observations.
Co-reporter:Joseph M. Beames ; Fang Liu ; Lu Lu
Journal of the American Chemical Society 2012 Volume 134(Issue 49) pp:20045-20048
Publication Date(Web):December 3, 2012
DOI:10.1021/ja310603j
Ozonolysis of alkenes in the troposphere produces Criegee intermediates, which have eluded detection in the gas phase until very recently. This laboratory has synthesized the simplest Criegee intermediate within a quartz capillary tube affixed to a pulsed valve to cool and isolate CH2OO in a supersonic expansion. UV excitation resonant with the B 1A′ ← X 1A′ transition depletes the ground-state population of CH2OO, which is detected by single-photon ionization at 118 nm. The large UV-induced depletion (approaching 100%) near the peak of the profile at 335 nm is indicative of rapid dissociation, consistent with the repulsive B 1A′ potential along the O–O coordinate computed theoretically. The experimental spectrum is in very good accord with the absorption spectrum calculated using the one-dimensional reflection principle. The B ← X spectrum is combined with the solar actinic flux to estimate an atmospheric lifetime for CH2OO at midday on the order of ∼1 s with respect to photodissociation.
Co-reporter:Pesia Soloveichik, Bridget A. O’Donnell and Marsha I. Lester, Joseph S. Francisco, Anne B. McCoy
The Journal of Physical Chemistry A 2010 Volume 114(Issue 3) pp:1529-1538
Publication Date(Web):October 15, 2009
DOI:10.1021/jp907885d
Infrared action spectroscopy is utilized to characterize the gas-phase, hydrogen-bonded H2O−HO complex, a primary interaction in the hydration of the hydroxyl radical. The OH radical stretch of the H2O−HO complex is identified at 3490 cm−1, shifted 78 cm−1 to lower frequency of the OH monomer transition. The stability of the complex, D0 ≤ 5.14 kcal mol−1, is derived from the highest observed OH product channel in the associated product state distribution. The assignment is supported by high level ab initio calculations of the spectral shift of the binary complex from free OH and its dissociation energy, De(CBS-∞) = 5.6 kcal mol−1. A second weaker feature, appearing 15 cm−1 to lower frequency at 3475 cm−1, is attributed to a hot band, the OH radical stretch originating from an out-of-plane H2O bending state, based on two-dimensional calculations of frequencies and strengths of transitions involving the coupled vibrational modes.
Co-reporter:Craig Murray, Erika L. Derro, Timothy D. Sechler and Marsha I. Lester
Accounts of Chemical Research 2009 Volume 42(Issue 3) pp:419
Publication Date(Web):December 29, 2008
DOI:10.1021/ar8001987
Weakly bound molecules—particularly hydrated complexes of abundant atmospheric species—have long been postulated to play an important role in atmospherically relevant reactions. For example, such complexes could seed cloud formation and alter the global radiation budget. In this Account, we initially describe the current data on weakly bound species produced in association reactions of the hydroxyl radical (OH) with molecular partners, particularly oxygen (O2), nitric acid (HONO2), and nitrogen dioxide (NO2). Researchers have identified weakly bound association products of these reactions as the hydrogen trioxy (HOOO) radical, the doubly hydrogen-bonded OH−HONO2 complex, and peroxynitrous acid (HOONO), respectively. In each case, previous kinetic studies of the reaction or OH vibrational relaxation processes have indicated unusual, non-Arrhenius behavior. Under the temperature−pressure conditions of the Earth’s lower atmosphere, these processes exhibit a negative temperature dependence, indicative of an attractive interaction, or a pressure dependence. Researchers have subsequently carried out extensive theoretical studies of the properties of these weakly bound molecules, but the theoretical studies have lacked experimental validation. Next, we describe experimental studies to determine the vibrational frequencies and stability of HOOO as a prototypical example of these weakly bound molecules. We then use these data to assess its importance in the atmosphere. We discuss the efficient production of the HOOO radical from OH and O2 under laboratory conditions and its subsequent detection using infrared action spectroscopy, a highly sensitive and selective double resonance technique. Using excitation of OH stretch and combination bands comprising OH stretch with lower frequency modes, we obtain detailed spectroscopic information on the vibrational modes of the two conformers of HOOO. In addition, we infer fundamental information about the dissociation dynamics from the OH product state distribution, which provides insight into the chemical bonding in HOOO. Perhaps most importantly, we utilize a simple conservation of energy relationship based on the highest energetically open OH product state to derive a rigorous upper limit for the stability of HOOO relative to the OH + O2 asymptote of 5.3 kcal mol−1. When combined with previous experimental rotational constants that reflect the structure of the HOOO radical, our laboratory characterization of its stability and vibrational frequencies provides critical information to assess its thermochemical properties. Using standard statistical mechanics approaches, we can calculate the likely atmospheric abundance of HOOO. We estimate that up to 25% of the OH radicals in the vicinity of the tropopause may be associated with O2 as a weakly bound molecule.
Co-reporter:Logan P. Dempsey, Timothy D. Sechler, Craig Murray and Marsha I. Lester
The Journal of Physical Chemistry A 2009 Volume 113(Issue 25) pp:6851-6858
Publication Date(Web):May 29, 2009
DOI:10.1021/jp902935c
The OH X2Π product state distribution arising from quenching of electronically excited OH A2Σ+ by O2 and CO2 under single collision conditions has been determined using a pump−probe technique. For both collision partners, the majority of OH X2Π products are observed in their lowest vibrational level, v′′ = 0, with significantly less population in v′′ = 1. The OH products from quenching by O2 are generated with a substantial degree of rotational excitation, peaking around N′′ ≈ 17, with an average rotational energy of ∼4800 cm−1, whereas OH products from quenching by CO2 exhibit a moderate degree of rotational excitation, peaking around N′′ ≈ 5, with an average rotational energy of ∼1800 cm−1. The branching fraction into OH X2Π products states reveals that nonreactive quenching is a significant decay pathway for both systems, accounting for at least 40(1)% of the quenched products with O2 and 64(5)% with CO2.
Co-reporter:Timothy D. Sechler, Logan P. Dempsey and Marsha I. Lester
The Journal of Physical Chemistry A 2009 Volume 113(Issue 31) pp:8845-8851
Publication Date(Web):July 15, 2009
DOI:10.1021/jp904978w
Vibrational energy transfer from v′ = 0 to v′ = 1 in the excited OH A2Σ+ electronic state is investigated in the collisional region of a free-jet expansion. Laser excitation is used to prepare high rotational levels in OH A2Σ+ (v′ = 0, N′ = 14−22), which lie above the energetic threshold for OH A2Σ+ (v′ = 1). Subsequent collisions with N2 result in population of a distribution of OH A2Σ+ (v′ = 1) product rotational levels that is characterized through dispersed fluorescence spectra. The majority of products are found in the most near-resonant rotational level of v′ = 1, with population falling off exponentially in lower rotational levels. Additionally, the efficiency of vibrational energy transfer is determined by comparing the emission from v′ = 1 products with that from the initially prepared v′ = 0 level. The fractional transfer decreases by an order of magnitude from the highest to lowest initial rotational levels investigated. This decrease is correlated with an increasingly large change in rotational angular momentum between the initial and final states. The results show that angular momentum constraints are the dominant factor in the efficiency of OH A2Σ+ v′ = 0 to v′ = 1 vibrational energy transfer at low collision energies.
Co-reporter:Logan P. Dempsey, Craig Murray, Patricia A. Cleary and Marsha I. Lester
Physical Chemistry Chemical Physics 2008 vol. 10(Issue 10) pp:1424-1432
Publication Date(Web):30 Nov 2007
DOI:10.1039/B715611A
A pump–probe laser-induced fluorescence technique has been used to examine the nascent OH X2Π product state distribution arising from non-reactive quenching of electronically excited OH A2Σ+ by molecular hydrogen and deuterium under single-collision conditions. The OH X2Π products were detected in v″ = 0, 1 and 2; the distribution peaks in v″ = 0 and decreases monotonically with increasing vibrational excitation. In all vibrational levels probed, the OH X2Π products are found to be highly rotationally excited, the distribution peaking at N″ = 15 when H2 was used as the collision partner and N″ = 17 for D2. A marked propensity for production of Π(A′) Λ-doublet levels was observed, while both OH X2Π spin–orbit manifolds were equally populated. These observations are interpreted as dynamical signatures of the nonadiabatic passage of the OH + H2/D2 system through the seams of conical intersection that couple the excited state (22A′) and ground state (12A′) surfaces.
Co-reporter:Erika L. Derro, Timothy D. Sechler, Craig Murray and Marsha I. Lester
The Journal of Physical Chemistry A 2008 Volume 112(Issue 39) pp:9269-9276
Publication Date(Web):May 29, 2008
DOI:10.1021/jp801232a
The DOOO radical has been produced by three-body association between OD and O2 in a supersonic free-jet expansion and investigated using action spectroscopy, an IR−UV double-resonance technique. Partially rotationally structured bands observed at 2635.06 and 5182.42 cm−1 are assigned to the OD stretch fundamental (νOD) and overtone (2νOD), respectively, of the trans-DOOO radical. Unstructured bands observed in both spectral regions are assigned to cis-DOOO. Nascent OD X2Π product state distributions following vibrational predissociation appear to be nearly statistical with respect to the degree of rotational excitation, but display a marked propensity for Π(A′) Λ-doublets, which is interpreted as a signature of a planar dissociation. The energetically highest open OD X2Π product channel implies an upper limit dissociation energy D0 ≤ 1856 cm−1 or 5.31 kcal mol−1. This value allows refinement of the upper limit D0 of the atmospherically important HOOO isotopomer, suggesting that it is marginally less stable than previously thought.
Co-reporter:Bridget A. O'Donnell;Eunice X. J. Li;Joseph S. Francisco;
Proceedings of the National Academy of Sciences 2008 105(35) pp:12678-12683
Publication Date(Web):August 4, 2008
DOI:10.1073/pnas.0800320105
Co-reporter:Erika L. Derro, Craig Murray, Marsha I. Lester and Mark D. Marshall
Physical Chemistry Chemical Physics 2007 vol. 9(Issue 2) pp:262-271
Publication Date(Web):20 Nov 2006
DOI:10.1039/B614152H
The photodissociation dynamics of methyl nitrate, CH3ONO2, has been investigated at 193 nm by examining the products from the primary dissociation channel, namely CH3O and NO2. The CH3O ( 2E) photoproducts were probed by laser-induced fluorescence (LIF) on the à 2A1– 2E transition under both nascent and jet-cooled conditions. The 310 and 311 bands originating from the vibrationless and C–O stretch (ν3) levels, respectively, were characterized to obtain the internal energy distribution of the CH3O products. Only a small fraction of the CH3O products (≤10%) were produced with one quantum of C–O stretch excitation as determined from the relative intensities of the bands in combination with transition probabilities derived from dispersed fluorescence measurements and/or calculated Franck–Condon factors. The CH3O products also had minimal rotational excitation: those produced in the ground vibrational state had a rotational temperature of 238 ± 7 K, corresponding to less than 1% of the available energy. Products with C–O stretch excitation were found to have a higher rotational temperature, but still a small fraction of the total energy. Combining the CH3O internal energy findings with previous photofragment translational energy measurements [X. Yang, P. Felder and J. R. Huber, J. Phys. Chem., 1993, 97, 10903] indicates that most of the available energy is deposited in the NO2 fragment. This is verified through dispersed fluorescence measurements which show that the NO2 fragment is produced electronically excited with internal energies extending to the NO + O dissociation limit. Ab initio calculations confirm that the dominant initial excitation is strongly localized on the NO2 moiety. The calculations are also used to reveal the forces that give rise to internal excitation of the CH3O fragment upon electronic excitation.
Co-reporter:Marsha I. Lester, Bethany V. Pond, Mark D. Marshall, David T. Anderson, Lawrence B. Harding and Albert F. Wagner
Faraday Discussions 2001 vol. 118() pp:373-385
Publication Date(Web):05 Jun 2001
DOI:10.1039/B009421H
A hydrogen-bonded complex composed of the OH and CO reactants has been identified along the OH + CO → HOCO reaction pathway. IR action spectroscopy in the OH overtone region has been used to examine the vibrational modes of the linear OH–CO complex, including intermolecular bending modes that probe portions of the reaction path leading to HOCO. The spectroscopic measurements have accessed highly excited intermolecular
levels, with energies up to 250 cm−1 above the zero-point level, which lie in close proximity to the transition state for reaction. The OH–CO binding energy, D0⩽430 cm−1, has also
been established from the quantum state distribution of the OH fragments following vibrational
predissociation of the OH–CO complex. Complementary electronic structure calculations have been performed to characterize the OH–CO and OH–OC complexes, the transition state for HOCO formation, and the direct reaction path that connects the experimentally
observed OH–CO complex to the HOCO intermediate.
Co-reporter:Fang Liu, Yi Fang, Manoj Kumar, Ward H. Thompson and Marsha I. Lester
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 32) pp:NaN20494-20494
Publication Date(Web):2015/07/22
DOI:10.1039/C5CP02917A
Many alkyl-substituted Criegee intermediates are predicted to undergo an intramolecular 1,4-hydrogen transfer to form isomeric vinyl hydroperoxide species (CCOOH moiety), which break apart to release OH and vinoxy radicals. We report direct detection of stabilized vinyl hydroperoxides formed via carboxylic acid-catalyzed tautomerization of Criegee intermediates. A doubly hydrogen-bonded interaction between the Criegee intermediate and carboxylic acid facilitates efficient hydrogen transfer through a double hydrogen shift. Deuteration of formic or acetic acid permits migration of a D atom to yield partially deuterated vinyl hydroperoxides, which are distinguished from the CH3CHOO, (CH3)2COO, and CH3CH2CHOO Criegee intermediates by mass. Using 10.5 eV photoionization, three prototypical vinyl hydroperoxides, CH2CHOOD, CH2C(CH3)OOD, and CH3CHCHOOD, are detected directly. Complementary electronic structure calculations reveal several reaction pathways, including the barrierless acid-catalyzed tautomerization reaction predicted previously and a barrierless addition reaction that yields hydroperoxy alkyl formate.
Co-reporter:Erika L. Derro, Craig Murray, Marsha I. Lester and Mark D. Marshall
Physical Chemistry Chemical Physics 2007 - vol. 9(Issue 2) pp:NaN271-271
Publication Date(Web):2006/11/20
DOI:10.1039/B614152H
The photodissociation dynamics of methyl nitrate, CH3ONO2, has been investigated at 193 nm by examining the products from the primary dissociation channel, namely CH3O and NO2. The CH3O ( 2E) photoproducts were probed by laser-induced fluorescence (LIF) on the à 2A1– 2E transition under both nascent and jet-cooled conditions. The 310 and 311 bands originating from the vibrationless and C–O stretch (ν3) levels, respectively, were characterized to obtain the internal energy distribution of the CH3O products. Only a small fraction of the CH3O products (≤10%) were produced with one quantum of C–O stretch excitation as determined from the relative intensities of the bands in combination with transition probabilities derived from dispersed fluorescence measurements and/or calculated Franck–Condon factors. The CH3O products also had minimal rotational excitation: those produced in the ground vibrational state had a rotational temperature of 238 ± 7 K, corresponding to less than 1% of the available energy. Products with C–O stretch excitation were found to have a higher rotational temperature, but still a small fraction of the total energy. Combining the CH3O internal energy findings with previous photofragment translational energy measurements [X. Yang, P. Felder and J. R. Huber, J. Phys. Chem., 1993, 97, 10903] indicates that most of the available energy is deposited in the NO2 fragment. This is verified through dispersed fluorescence measurements which show that the NO2 fragment is produced electronically excited with internal energies extending to the NO + O dissociation limit. Ab initio calculations confirm that the dominant initial excitation is strongly localized on the NO2 moiety. The calculations are also used to reveal the forces that give rise to internal excitation of the CH3O fragment upon electronic excitation.
Co-reporter:Logan P. Dempsey, Craig Murray, Patricia A. Cleary and Marsha I. Lester
Physical Chemistry Chemical Physics 2008 - vol. 10(Issue 10) pp:NaN1432-1432
Publication Date(Web):2007/11/30
DOI:10.1039/B715611A
A pump–probe laser-induced fluorescence technique has been used to examine the nascent OH X2Π product state distribution arising from non-reactive quenching of electronically excited OH A2Σ+ by molecular hydrogen and deuterium under single-collision conditions. The OH X2Π products were detected in v″ = 0, 1 and 2; the distribution peaks in v″ = 0 and decreases monotonically with increasing vibrational excitation. In all vibrational levels probed, the OH X2Π products are found to be highly rotationally excited, the distribution peaking at N″ = 15 when H2 was used as the collision partner and N″ = 17 for D2. A marked propensity for production of Π(A′) Λ-doublet levels was observed, while both OH X2Π spin–orbit manifolds were equally populated. These observations are interpreted as dynamical signatures of the nonadiabatic passage of the OH + H2/D2 system through the seams of conical intersection that couple the excited state (22A′) and ground state (12A′) surfaces.
.jpg)