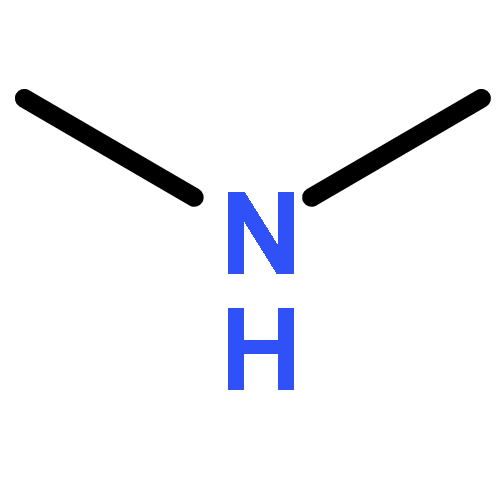
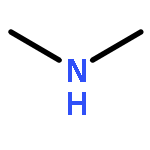
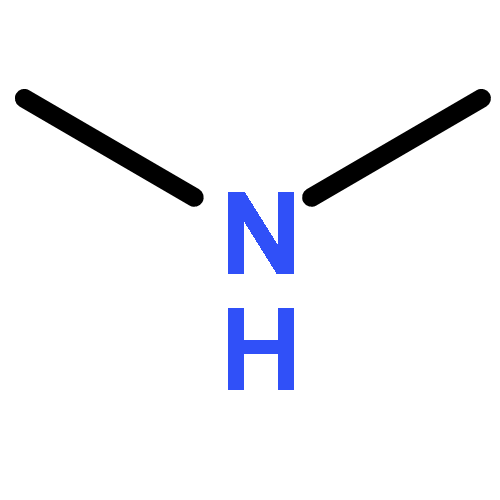
Co-reporter:Ziran Liu, Bin Peng, Qiong Luo, Qian-shu Li, Yaoming Xie, R. Bruce King
Journal of Fluorine Chemistry 2014 Volume 157() pp:22-29
Publication Date(Web):January 2014
DOI:10.1016/j.jfluchem.2013.10.018
•The species (CF3NC)2Co2(CO)n (n = 7, 6, 5, 4) have been studied by density functional theory.•Bridging CF3NC groups are energetically preferred over bridging CO groups.•Four-electron donor bridging η2-μ-CF3NC groups are found in higher energy (CF3NC)2Co2(CO)n (n = 5, 4) structures.•Coupling of two CF3NC groups to form a CF3NCCNCF3 ligand occurs in the (CF3NC)2Co2(CO)7 system.The cobalt carbonyl complexes (CF3NC)2Co2(CO)n (n = 7, 6, 5, 4) and (CF3NC)Co(CO)n(n = 4, 3, 2) of the strongly π-accepting trifluoromethyl isocyanide ligand have been investigated by density functional theory. The lowest energy (CF3NC)2Co2(CO)6 structures are doubly bridged structures with the global minimum having two bridging CF3NC ligands. The lowest energy unbridged (CF3NC)2Co2(CO)6 structure lies ∼19 kcal/mol in energy above this global minimum. This differs from unsubstituted Co2(CO)8 for which the doubly bridged and unbridged isomers have similar energies so both can be observed experimentally. The lowest energy structures of the formally unsaturated (CF3NC)2Co2(CO)n (n = 5, 4) derivatives are also doubly bridged structures. Bridging CF3NC groups are energetically preferred over bridging CO groups. Four-electron donor bridging η2-μ-CF3NC groups are found in higher energy structures. Coupling of CF3NC groups to form a coordinated bis(trifluoromethyldiimine) ligand, CF3NCCNCF3 is observed in carbonyl-rich (CF3NC)2Co2(CO)7 structures. However, such (CF3NC)2Co2(CO)7 structures do not appear to be viable since CO dissociation from the lowest energy such structure to give (CF3NC)2Co2(CO)6 is predicted to be essentially thermoneutral.The lowest energy (CF3NC)2Co2(CO)6 structures are doubly bridged structures with the global minimum having two bridging CF3NC ligands. The lowest energy structures of the formally unsaturated (CF3NC)2Co2(CO)n (n = 5, 4) derivatives are also doubly bridged structures with bridging CF3NC groups being energetically preferred over bridging CO groups. Four-electron donor bridging η2-μ-CF3NC groups are found in higher energy structures.
Co-reporter:Yan Wu, Qiong Luo, Chaoyang Wang, Qian-shu Li, Yaoming Xie, R. Bruce King
Inorganica Chimica Acta 2013 Volume 394() pp:322-327
Publication Date(Web):1 January 2013
DOI:10.1016/j.ica.2012.07.017
The binuclear iron carbonyl derivatives Fe2(CH2)(CO)8 and Fe2(CF2)(CO)8 are known compounds that have been synthesized and characterized structurally by X-ray crystallography. Their low-lying structures have now been investigated using density functional theory. The C2v isomers of Fe2(CH2)(CO)8 with triply and singly bridged Fe–Fe bonds are found to have closely spaced energies. These observations are consistent with the experimental observation of an equilibrium between the Fe2(CH2)(CO)8 isomer with two bridging CO groups and the isomer with all terminal CO groups. The Fe2(CH2)(CO)8 system appears to be analogous to the well-documented Co2(CO)8 system, where an equilibrium between doubly bridged and unbridged isomers is observed experimentally under ambient conditions. In addition to these two low energy Fe2(CH2)(CO)8 structures, a higher energy structure is found in which one of the CO groups has inserted into an Fe–CH2 bond to give a bridging ketene (CH2CO) ligand. The lowest-lying structure for the corresponding fluorinated derivative Fe2(CF2)(CO)8 is the C2v isomer with two symmetrical bridging CO groups as well as a bridging CF2 group. The corresponding Fe2(CF2)(CO)8 isomer with all terminal CO groups, also of C2v symmetry, is also a low-energy structure. In addition to these two low-energy Fe2(CF2)(CO)8 structures with a bridging CF2 group, two higher energy structures are found with terminal CF2 groups. The higher energies of ∼15 kcal/mol above the lowest energy structure for these terminal CF2 structures suggest that CF2 groups prefer to be bridging rather than terminal ligands in binuclear metal derivatives. No corresponding Fe2(CH2)(CO)8 structures with terminal CH2 groups were found at accessible energies relative to the lowest energy structure.Graphical abstractThe lowest energy Fe2(CX2)(CO)8 (X = H, F) structures have bridging CX2 groups and either two bridging CO groups or all terminal CO groups. Equilibrium mixtures of both types of isomers are predicted in accord with experiment. A higher energy Fe2(CH2)(CO)8 structure has a bridging ketene ligand. Higher energy Fe2(CF2)(CO)8 structures are found with terminal CF2 ligands.Highlights► The energetically low-lying structures of CX2Fe2(CO)8 (X = H, F) have been characterized by density functional theory. ► CH2Fe2(CO)8 has isomers with and without bridging CO groups in equilibrium similar to Co2(CO)8. ► A higher energy (CH2CO)Fe2(CO)7 structure is found with a bridging ketene ligand. ► Terminal CF2 groups are found in higher energy CF2Fe2(CO)8 structures.
Co-reporter:Lihong Tang, Qiong Luo, Qian-shu Li, Yaoming Xie, R. Bruce King, and Henry F. Schaefer III
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 3) pp:862-874
Publication Date(Web):January 30, 2012
DOI:10.1021/ct200820v
The synthesis by Power and co-workers of the first metal–metal quintuple bond (Science2005, 310, 844) is a landmark in inorganic chemistry. The 18-electron rule suggests that Nb2(CO)9 and Nb2(CO)8 are candidates for binary metal carbonyls containing metal–metal quadruple and quintuple bonds, respectively. Density functional theory (MPW1PW91 and BP86) indeed predicts structures having very short Nb–Nb distances of ∼2.5 Å for Nb2(CO)9 and ∼2.4 Å for Nb2(CO)8 as well as relatively large Nb–Nb Wiberg bond indices supporting these high formal Nb–Nb bond orders. However, analysis of the frontier molecular orbitals of these unbridged structures suggests formal Nb≡Nb triple bonds and 16-electron metal configurations. This contrasts with an analysis of the frontier orbitals in a model chromium(I) alkyl linear CH3CrCrCH3, which confirms the generally accepted presence of chromium–chromium quintuple bonds in such molecules. The presence of Nb≡Nb triple bonds rather than quadruple or quintuple bonds in the Nb2(CO)n (n = 9, 8) structures frees up d(xy) and d(x2–y2) orbitals for dπ→pπ* back-bonding to the carbonyl groups. The lowest energy Nb2(CO)n structures (n = 9, 8) are not these unbridged structures but structures having bridging carbonyl groups of various types and formal Nb–Nb orders no higher than three. Thus, the two lowest energy Nb2(CO)9 structures have Nb≡Nb triple bond distances of ∼2.8 Å and three semibridging carbonyl groups, leading to a 16-electron configuration rather than an 18-electron configuration for one of the niobium atoms. The lowest energy structure of the highly unsaturated Nb2(CO)8 is unusual since it has a formal single Nb–Nb bond of length ∼3.1 Å and two four-electron donor η2-μ-CO groups, thereby giving each niobium atom only a 16-electron configuration.
Co-reporter:Qiong Luo
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 43) pp:14878-14885
Publication Date(Web):30 Jul 2012
DOI:10.1039/C2CP41820G
The neutral carbon aluminium cluster Al3C and its anion Al3C− have been investigated at the B3LYP/6-311+G(d) and BP86/6-311+G(d) levels of theory. It is suggested that the C2v neutral cluster Al3C could be considered as a pseudohalogen superatom with a smaller electron affinity than the iodine atom. The DFT calculation results confirm that, similar to the halogen atoms, the pseudohalogen superatom Al3C cluster could form the compounds corresponding to the dimer, salts, interhalogen compounds, oxides, acid radicals, and coordination complexes as well as superatom compounds of halogen, showing that Al3C has similar chemical properties to halogens and maintains its integrity in the related reactions. Based on the maintenance of integrity in the chemical assemblies [(Al3C)(KCAl3)n]− (n = 1–5), it could be anticipated that the neutral cluster Al3C holds great potential as a building block for the development of future nanostructured materials. Further, corresponding experimental verifications are invited.
Co-reporter:Yan Hong Liang, Qiong Luo, Min Guo and Qian Shu Li
Dalton Transactions 2012 vol. 41(Issue 39) pp:12075-12081
Publication Date(Web):01 Aug 2012
DOI:10.1039/C2DT31016C
A series of polynitrogen molecules composed by multiple cyclic-N3 and/or cyclic-N5 are systematically investigated by using DFT and MP2 methods. It is found that all energetically favored structures appear with a corner to corner connection between two rings for the cyclic-N3 and/or the cyclic-N5. Furthermore, straight interaction between the N3-rings would decrease the stability of cyclic-N3 relative to the cyclic-N3 radical and thus not favor the formation of a stable structure containing multiple cyclic-N3, while the interaction between the N5-rings can improve the stability of cyclic-N5 and tend to produce a stable structure composed by multiple cyclic-N5. For the stable structure consisting of cyclic-N3 and cyclic-N5, the charge is transferred from cyclic-N3 to cyclic-N5, facilitating the stability of N5, and the attacked site of the structure by the radicals is analyzed by condensed Fukui function. Finally, the stabilities of the structures N3(N5)3 and N5(N5)5 are investigated and demonstrated to be attributed to the charge transfer and the coordination number of N3-ring and N5-ring.
Co-reporter:Lihong Tang, Qiong Luo, Qian-shu Li, Yaoming Xie, R. Bruce King, and Henry F. Schaefer III
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 7) pp:2112-2125
Publication Date(Web):May 31, 2011
DOI:10.1021/ct2003513
The dissociation of Nb2(CO)12 into two Nb(CO)6 units is predicted to require ∼13 kcal/mol so that Nb2(CO)12 rather than Nb(CO)6 is the anticipated initial oxidation product of the known Nb(CO)6– anion. This differs from the corresponding vanadium carbonyl chemistry where V(CO)6 rather than V2(CO)12 is found experimentally to be the oxidation product of V(CO)6–. The lowest energy Nb2(CO)12 structure consists of two Nb(CO)6 fragments joined by a Nb–Nb bond of ∼3.4 Å length so that each niobium atom is heptacoordinate, counting the metal–metal bond. These niobium coordination polyhedra can be approximated by capped octahedra. Among unsaturated binuclear niobium carbonyls the lowest energy Nb2(CO)11 structure has a formal four-electron donor bridging η2-μ-CO group and a formal Nb–Nb single bond rather than only two-electron donor carbonyl groups and a formal Nb═Nb double bond. The Nb2(CO)11 structures with formal Nb═Nb double bonds and exclusively two-electron donor carbonyl groups lie more than 13 kcal/mol above this low-energy Nb2(CO)10(η2-μ-CO) structure. However, Nb2(CO)11 is predicted to be thermodynamically disfavored, owing to disproportionation into Nb2(CO)12 + Nb2(CO)10, a slightly exothermic process by ∼4 kcal/mol. The Nb2(CO)10 structures with formal Nb≡Nb triple bonds and all two-electron donor carbonyl groups appear to be particularly favorable, as suggested by high CO dissociation energies and viability toward disproportionation. Such structures are isolobal with Cp2Mo2(CO)4, which was the first stable metal carbonyl to be discovered with a short metal–metal distance, corresponding to a formal triple bond. Considerably higher energy Nb2(CO)10 structures (by more than 20 kcal/mol) have two four-electron donor bridging carbonyl groups and long niobium–niobium distances. Such structures can be considered to consist of a bidentate Nb(CO)6 “ligand” coordinating to a Nb(CO)4 unit through the two η2-μ-CO groups.
Co-reporter:Qiong Luo
Science China Chemistry 2008 Volume 51( Issue 7) pp:607-613
Publication Date(Web):2008 July
DOI:10.1007/s11426-008-0073-9
For a series of boron rings with planar hyper-coordinate 8th group transition metal atoms, singlet 1FeB8−2, multiplet kFeB9n (n = −1, k = 1; n = 0, k = 2), singlet 1CoB8n(n = −1, +1, +3), multiplet kCoB9n (n = +1, k = 2; n = 0, k = 1) and singlet 1NiB9+, the geometry structures have been optimized to be local minima on corresponding potential hyper-surfaces. The electron structures are discussed by orbital analysis and the aromaticity is predicted by nucleus-independent chemical shifts calculation at both the B3LYP/6-311+G* and BP86/6-311+G* levels of theory, respectively. The results suggest that all these structures with high symmetry planar geometries are stable and have aromatic properties with six π valence electrons.
Co-reporter:Qiong Luo
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 43) pp:NaN14885-14885
Publication Date(Web):2012/07/30
DOI:10.1039/C2CP41820G
The neutral carbon aluminium cluster Al3C and its anion Al3C− have been investigated at the B3LYP/6-311+G(d) and BP86/6-311+G(d) levels of theory. It is suggested that the C2v neutral cluster Al3C could be considered as a pseudohalogen superatom with a smaller electron affinity than the iodine atom. The DFT calculation results confirm that, similar to the halogen atoms, the pseudohalogen superatom Al3C cluster could form the compounds corresponding to the dimer, salts, interhalogen compounds, oxides, acid radicals, and coordination complexes as well as superatom compounds of halogen, showing that Al3C has similar chemical properties to halogens and maintains its integrity in the related reactions. Based on the maintenance of integrity in the chemical assemblies [(Al3C)(KCAl3)n]− (n = 1–5), it could be anticipated that the neutral cluster Al3C holds great potential as a building block for the development of future nanostructured materials. Further, corresponding experimental verifications are invited.
Co-reporter:Yan Hong Liang, Qiong Luo, Min Guo and Qian Shu Li
Dalton Transactions 2012 - vol. 41(Issue 39) pp:NaN12081-12081
Publication Date(Web):2012/08/01
DOI:10.1039/C2DT31016C
A series of polynitrogen molecules composed by multiple cyclic-N3 and/or cyclic-N5 are systematically investigated by using DFT and MP2 methods. It is found that all energetically favored structures appear with a corner to corner connection between two rings for the cyclic-N3 and/or the cyclic-N5. Furthermore, straight interaction between the N3-rings would decrease the stability of cyclic-N3 relative to the cyclic-N3 radical and thus not favor the formation of a stable structure containing multiple cyclic-N3, while the interaction between the N5-rings can improve the stability of cyclic-N5 and tend to produce a stable structure composed by multiple cyclic-N5. For the stable structure consisting of cyclic-N3 and cyclic-N5, the charge is transferred from cyclic-N3 to cyclic-N5, facilitating the stability of N5, and the attacked site of the structure by the radicals is analyzed by condensed Fukui function. Finally, the stabilities of the structures N3(N5)3 and N5(N5)5 are investigated and demonstrated to be attributed to the charge transfer and the coordination number of N3-ring and N5-ring.