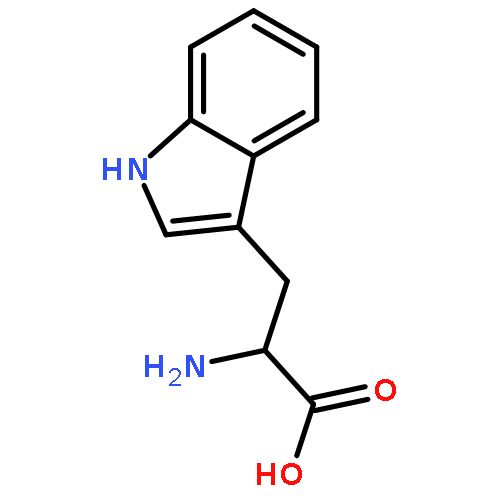
Co-reporter:Guillem Valero
Chirality 2016 Volume 28( Issue 8) pp:599-605
Publication Date(Web):
DOI:10.1002/chir.22618
Abstract
Catalysis of the aldol reaction between cyclohexanone and 4-nitrobenzaldehyde by mixtures of L-Arg and of L-Glu in wet dimethyl sulfoxide (DMSO) takes place with higher enantioselectivity (up to a 7-fold enhancement in the anti-aldol for the 1:1 mixture) than that observed when either L-Glu or L-Arg alone are used as the catalysts. These results can be explained by the formation of a catalytically active hydrogen-bonded complex between both amino acids, and demonstrate the possibility of positive cooperative effects in catalysis by two different α-amino acids. Chirality 28:599–605, 2016. © 2016 Wiley Periodicals, Inc.
Co-reporter:Guillem Valero;Dr. Josep M. Ribó ;Dr. Albert Moyano
Chemistry - A European Journal 2014 Volume 20( Issue 52) pp:17395-17408
Publication Date(Web):
DOI:10.1002/chem.201404497
Abstract
The aldol reaction between acetone and 4-nitrobenzaldehyde run in the nominal absence of any enantioselective catalyst was monitored by chiral HPLC with the aid of an internal standard. The collected data show the presence of a detectable initial enantiomeric excess of the aldol product in the early stages of the reaction in about 50 % of the experiments. Only a small fraction of the reaction contained the non-racemic aldol product after 24 h. This temporary emergence of natural optical activity could be the signature of a coupled reaction network that leads to a spontaneous mirror-symmetry-breaking process, which originates at very low conversions (i.e., strongly depends on events taking place at the very first stages of the process). The reaction is not autocatalytic in the aldol product, which rules out a simple Frank-type reaction network as the source of the observed symmetry breaking. On the other hand, the isolation and characterisation of a double-aldol adduct suggested a reaction network that involved both indirect autocatalysis and indirect mutual inhibition between the enantiomers of the reaction product.
Co-reporter:Juliana C. Gomes, Jordi Sirvent, Albert Moyano, Manoel T. Rodrigues Jr., and Fernando Coelho
Organic Letters 2013 Volume 15(Issue 22) pp:5838-5841
Publication Date(Web):November 4, 2013
DOI:10.1021/ol4029034
The readily available bicyclic imidazolyl alcohol 1 is a unique catalyst for the aqueous Morita–Baylis–Hillman (MBH) reaction between unprotected isatins and cyclic enones that gives access to a variety of potentially very useful 3-substituted 3-hydroxy-2-oxindoles in an operationally simple, efficient, and environmentally friendly way. The hydroxyl group of the catalyst is believed to stabilize the betaine intermediate formed in the first step of the MBH reaction.
Co-reporter:Ana Pou
European Journal of Organic Chemistry 2013 Volume 2013( Issue 15) pp:3103-3111
Publication Date(Web):
DOI:10.1002/ejoc.201300197
Abstract
α-Branched α,β-unsaturated aldehydes have been tested in the organocatalytic tandem Michael addition/cyclization with N-(benzyloxycarbonyl)hydroxylamine, a reaction which until now has been restricted to α-unsubstituted enals. Starting from cyclopentene-2-carbaldehyde, and using diphenylprolinol trimethylsilyl ether as a chiral amine catalyst, this approach has led to the development of a practical, high yielding (93–98 % overall yield, three steps), and highly enantioselective (up to 98:2 er) route to the cyclic β-amino acid cispentacin, which compares favourably with previously described asymmetric syntheses of this biologically active natural product. When using acyclic α-branched α,β-unsaturated aldehydes as substrates, the reaction yields depend on the substitution pattern of the aldehydes, and mixtures of cis- and trans-isomers are obtained. Nevertheless, this strategy has proved to be successful in some instances, and (3R,4R)-benzyl 3-ethyl-4-methyl-5-oxoisoxazolidin-2-carboxylate could be obtained in 70 % overall yield (two steps) from the reaction of 2-ethylcrotonaldehyde and N-(benzyloxycarbonyl)hydroxylamine under catalysis with diphenylprolinol trimethylsilyl ether, and with high enantiomeric purity (99:1 er).
Co-reporter:Xavier Companyó, Guillem Valero, Oriol Pineda, Teresa Calvet, Mercè Font-Bardía, Albert Moyano and Ramon Rios
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 2) pp:431-439
Publication Date(Web):06 Oct 2011
DOI:10.1039/C1OB06503C
An enantioselective α-oxyamination of unprotected 3-substituted oxindoles with nitrosobenzene catalyzed by tertiary amine–thiourea bifunctional organocatalysts has been developed and affords the corresponding 3-amino-2-oxindole derivatives in good yields and with moderate to excellent enantioselectivities (up to > 99.9:0.1 er when the product is isolated by direct filtration from the reaction mixture). The absolute configuration of the major enantiomers of the products has been established both by chemical correlation and by comparison between the theoretically calculated and the experimental ECD.
Co-reporter:Andrea-Nekane R. Alba, Guillem Valero, Teresa Calbet, Mercé Font-Bardía, Albert Moyano and Ramon Rios
New Journal of Chemistry 2012 vol. 36(Issue 3) pp:613-618
Publication Date(Web):28 Nov 2011
DOI:10.1039/C1NJ20659A
The catalytic enantioselective addition of 2-tert-butyl-4-aryl-1,3-oxazol-5-ones to maleimides is reported. The addition takes place exclusively at the C-4 position of the oxazolone ring, giving access to quaternary amino acid derivatives. The reaction is catalyzed by readily available chiral bases such as (DHQD)2PYR, rendering the final compounds in good yields and with moderate to good diastereo- and enantioselectivities. When the C-4-substituent of the 2-tert-butyl-oxazolone is an alkyl group, the regioisomeric C-2 addition product is also obtained.
Co-reporter:Albert Moyano and Ramon Rios
Chemical Reviews 2011 Volume 111(Issue 8) pp:4703
Publication Date(Web):May 26, 2011
DOI:10.1021/cr100348t
Co-reporter:Xavier Companyó, Guillem Valero, Victor Ceban, Teresa Calvet, Mercé Font-Bardía, Albert Moyano and Ramon Rios
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 23) pp:7986-7989
Publication Date(Web):13 Sep 2011
DOI:10.1039/C1OB06308A
The highly enantioselective asymmetric allylic alkylation of Morita–Baylis–Hillman carbonates with bis(phenylsulfonyl)methane is presented. The reaction is simply catalyzed by cinchona alkaloid derivatives affording the final alkylated products in good yields and enantioselectivities.
Co-reporter:Alex Zea, Andrea-Nekane R. Alba, Natalia Bravo, Albert Moyano, Ramon Rios
Tetrahedron 2011 67(14) pp: 2513-2529
Publication Date(Web):
DOI:10.1016/j.tet.2011.02.032
Co-reporter:Alex Zea;Guillem Valero;Andrea-NekaneR. Alba;Ramon Rios
Advanced Synthesis & Catalysis 2010 Volume 352( Issue 7) pp:1102-1106
Publication Date(Web):
DOI:10.1002/adsc.201000031
Abstract
For the first time, the addition of anthrones to maleimides catalyzed by bifunctional thiourea catalysts is reported. The thiourea moiety is able to activate the maleimide and the tertiary amine deprotonates the anthrone, furnishing the final Diels–Alder or Michael adducts in excellent yields and enantioselectivities.
Co-reporter:Martin Kamlar;Natalia Bravo;Andrea-Nekane R. Alba;Simona Hybelbauerová;Ivana Císa&x159;ová;Jan Veselý;Ramon Rios
European Journal of Organic Chemistry 2010 Volume 2010( Issue 28) pp:5464-5470
Publication Date(Web):
DOI:10.1002/ejoc.201000851
Abstract
An organocatalytic, highly enantioselective addition of 1-fluoro-1-nitro(phenylsulfonyl)methane to α,β-unsaturated aldehydes is reported. The reaction is simply catalyzed by secondary amines and furnishes the corresponding fluorinated derivatives in good yields, with moderate diastereoselectivities and excellent enantioselectivities. The absolute configuration of the major diastereomers was unambiguously ascertained by X-ray diffraction analysis.
Co-reporter:Natalia Bravo, Andrea-Nekane R. Alba, Guillem Valero, Xavier Companyó, Albert Moyano and Ramon Rios
New Journal of Chemistry 2010 vol. 34(Issue 9) pp:1816-1820
Publication Date(Web):22 Jun 2010
DOI:10.1039/C0NJ00321B
Azlactones react with 1,2-bis(phenylsulfonyl)ethene under catalysis by simple chiral thioureas, affording α,α-disubstituted α-amino acid derivatives in good yields and in moderate to good enantioselectivities.
Co-reporter:Andrea-NekaneR. Alba;Guillem Valero;Teresa Calbet;Mercé Font-Bardía Dr.;Ramon Rios Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 32) pp:9884-9889
Publication Date(Web):
DOI:10.1002/chem.201000239
Abstract
The first highly diastereo- and enantioselective organocatalytic synthesis of 2,2-disubstituted-2H-oxazol-5-ones is described. The addition of oxazolones to maleimides is promoted by bifunctional thiourea catalysts, which afford the corresponding 2,2-disubstituted-2H-oxazol-5-ones with total regio- and stereocontrol.
Co-reporter:Andrea-NekaneR. Alba;Xavier Companyó;Guillem Valero, Dr. ;Ramon Rios Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 18) pp:5354-5361
Publication Date(Web):
DOI:10.1002/chem.200903025
Abstract
A new, easy, and highly enantioselective method for the synthesis of quaternary α-alkyl-α-amino acids based on organocatalysis is reported. The addition of oxazolones to 1,1-bis(phenylsulfonyl)ethylene is efficiently catalyzed by simple chiral bases or thioureas. The reaction affords α,α-disubstituted α-amino acid derivatives with complete C4 regioselectivity and with excellent yields and enantioselectivities. This methodology is complementary to previously reported enantioselective approaches to quaternary α-amino acids and allows the synthesis of α-phenyl-α-alkyl-α-amino acids and α-tert-butyl-α-alkyl-α-amino acids. It has distinct advantages in terms of operational simplicity, enviromentally friendly conditions, and suitability for large-scale reactions.
Co-reporter:Niama El-Hamdouni;Xavier Companyó;Ramon Rios Dr. Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 4) pp:1142-1148
Publication Date(Web):
DOI:10.1002/chem.200902678
Co-reporter:Albert Moyano ;Niama El-Hamdouni;Ahmed Atlamsani
Chemistry - A European Journal 2010 Volume 16( Issue 18) pp:5260-5273
Publication Date(Web):
DOI:10.1002/chem.200903410
Abstract
Rearrangement reactions often lead to the regio- and stereoselective formation of carbon–carbon or carbon–heteroatom bonds, and allow the construction of otherwise hard-to-access molecular frameworks. Research disclosed in the present decade, especially in the last two years, has shown that organocatalytic modes of activation can be successfully applied to a variety of rearrangements. In this Minireview we discuss the advances achieved so far in asymmetric organocatalytic rearrangement reactions.
Co-reporter:Laura Mendieta;Anna Picó Dr.;Teresa Tarragó Dr.;Meritxell Teixidó Dr.;Marcos Castillo Dr.;Llorenç Rafecas Dr.;Ernest Giralt Dr.
ChemMedChem 2010 Volume 5( Issue 9) pp:1556-1567
Publication Date(Web):
DOI:10.1002/cmdc.201000109
Abstract
Herein we present the design, synthesis, and evaluation of a structurally novel library of 20 peptidyl 3-aryl vinyl sulfones as inhibitors of cathepsins L and B. The building blocks, described here for the first time, were synthesized in a highly efficient and enantioselective manner, starting from 3-aryl-substituted allyl alcohols. The corresponding vinyl sulfones were prepared by a new approach, based on a combination of solid-phase peptide synthesis using the Fmoc/tBu strategy, followed by solution-phase coupling to the corresponding (R)-3-amino-3-aryl vinyl sulfones as trifluoroacetate salts. The inhibitory activity of the resulting compounds against cathepsins L and B was evaluated, and the compound exhibiting the best activity was selected for enzymatic characterization. Finally, docking studies were performed in order to identify key structural features of the aryl substituent.
Co-reporter:Andrea-Nekane Balaguer;Xavier Companyó;Teresa Calvet;Mercé Font-Bardía;Ramon Rios
European Journal of Organic Chemistry 2009 Volume 2009( Issue 2) pp:199-203
Publication Date(Web):
DOI:10.1002/ejoc.200801005
Abstract
A convenient and novel oxazol-5-one addition to nitrostyrenes is reported. The reaction is catalyzed by tertiary amines and yields the corresponding adducts with total regio- and diastereoselectivity. The addition exclusively takes place at the C-2 position of oxazol-5-ones, furnishing diastereopure N,O-aminals. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Andrea-Nekane Alba, Pilar Gómez-Sal, Ramon Rios, Albert Moyano
Tetrahedron: Asymmetry 2009 Volume 20(Issue 11) pp:1314-1318
Publication Date(Web):19 June 2009
DOI:10.1016/j.tetasy.2009.05.011
The proline-catalyzed aldol reaction of racemic 2-(2′-pyrimidyl)ferrocenecarbaldehyde with acetone in DMSO at room temperature constitutes as the first example of an organocatalytic kinetic resolution of a planar-chiral compound. The selectivity factor of the kinetic resolution is 9.2, and the stereochemical outcome of the process can be easily rationalized by the standard mechanistic model of the proline-catalyzed aldol reaction.(pR)-2-(2′-Pyrimidyl)ferrocenecarbaldehydeC15H12FeN2OEe = 78% (HPLC)[α]D20=+26.0 (c 1.1, CHCl3)Source of chirality: organocatalytic kinetic resolutionAbsolute configuration: (pR)(pS,E)-4-[2-(2′-Pyrimidyl)ferrocenyl]but-3-ene-2-oneC18H16FeN2OEe = 56% (HPLC)[α]D20=+648 (c 0.05, CHCl3)Source of chirality: organocatalytic kinetic resolutionAbsolute configuration: (pS,E)(R,pS)-4-[2-(2′-Pyrimidyl)ferrocenyl]-4-hydroxy-2-butanoneC18H18FeN2O2Ee = 52% (HPLC)[α]D20=-28.0 (c 0.05, CHCl3)Source of chirality: organocatalytic kinetic resolutionAbsolute configuration: (R,pS)
Co-reporter:Andrea-Nekane Alba;Xavier Companyó Dr.;Ramon Rios Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 42) pp:11095-11099
Publication Date(Web):
DOI:10.1002/chem.200901806
Co-reporter:Xavier Companyó;Guillem Valero;Luis Crovetto Dr. Dr.;Ramon Rios Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 27) pp:6564-6568
Publication Date(Web):
DOI:10.1002/chem.200900488
Co-reporter:Natalia Bravo, Ignasi Mon, Xavier Companyó, Andrea-Nekane Alba, Albert Moyano, Ramon Rios
Tetrahedron Letters 2009 50(48) pp: 6624-6626
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.09.038
Co-reporter:Andrea-Nekane Alba, Natalia Bravo, Albert Moyano, Ramon Rios
Tetrahedron Letters 2009 50(25) pp: 3067-3069
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.04.037
Co-reporter:Xavier Companyó, Monika Hejnová, Martin Kamlar, Jan Vesely, Albert Moyano, Ramon Rios
Tetrahedron Letters 2009 50(35) pp: 5021-5024
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.06.092
Co-reporter:Guillem Valero, Jiri Schimer, Ivana Cisarova, Jan Vesely, Albert Moyano, Ramon Rios
Tetrahedron Letters 2009 50(17) pp: 1943-1946
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.02.049
Co-reporter:Rosa M. Moreno;Mònica Catasús;Concepción López
European Journal of Organic Chemistry 2008 Volume 2008( Issue 14) pp:2388-2396
Publication Date(Web):
DOI:10.1002/ejoc.200800029
Abstract
An enantiocontrolled synthesis of α-ferrocenylalanine methyl ester (S)-24 and of the corresponding N-Boc derivative (S)-25 is presented. We have found that an important source of instability for α-ferrocenyl-α-amino acid derivatives is the high reactivity of the α-hydrogen, and its replacement by a methyl group has proven to be essential for the stability of these compounds. The synthesis of (S)-24 requires only six steps from the readily available 2-propenylferrocene which is converted into the diol (S)-16 by Sharpless catalytic dihydroxylation. Regio- and stereoselective azide substitution of the tertiary hydroxy group affords the azido diol (S)-17. Swern oxidation gives the corresponding aldehyde which is smoothly oxidized to the carboxylic ester by means of the Smith–Kozlowski protocol. Transesterification and azide reduction lead to the α-amino ester (S)-24 in a good overall yield and enantiomeric purity. A cyclic voltammetric study of this compound showed that oxidation takes place by a simple reversible one-electron-transfer process. Half a century after the initial unsuccessful attempts to obtain ferrocenylglycine, this is the first synthesis of a stable α-amino acid derivative incorporating a ferrocene unit at the α-position.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2008)
Co-reporter:Agustí Bueno;Malgorzata Rosol;Jasón García
Advanced Synthesis & Catalysis 2006 Volume 348(Issue 18) pp:
Publication Date(Web):15 DEC 2006
DOI:10.1002/adsc.200600282
The first non-enzymatic kinetic resolution of planar chiral ferrocenes has been achieved by the Sharpless catalytic asymmetric dihydroxylation (AD) of a set of racemic 2-substituted 1-ethenylferrocenes 1a–d. The enantioselectivity factor krel varies from 20 to 62 [for (DHQD)2PYR ligands], and from 5 to 27 [for (DHQ)2PYR ligands]. The stereochemical outcome of the resolution can be easily predicted by the mnemonic device for AD, with the additional hypothesis that in the preferred transition state the olefin group and the upper cyclopentadiene ring of vinylferrocenes exhibit an essentially coplanar geometry.
Co-reporter:Rosa M Moreno, Albert Moyano
Tetrahedron: Asymmetry 2006 Volume 17(Issue 7) pp:1104-1110
Publication Date(Web):3 April 2006
DOI:10.1016/j.tetasy.2006.03.017
Chiral Schiff bases arising from the condensation of a set of diversely substituted (S)-2-amino-2-ferrocenyl ethanol derivatives 1a–e with salicylaldehydes 2A–B have been tested in the asymmetric, titanium-promoted diketene addition to benzaldehyde. This study has revealed that the enantiofacial selectivity of this reaction depends strongly on the substitution pattern of the amino alcohol component. Molecular mechanics calculations have led to the identification of a transition state model that explains these observations.Chiral Schiff base ligands derived from salicylaldehydes and from a set of diversely substituted (S)-2-amino-2-ferrocenylethanols show an unprecedented stereodivergence in the titanium-promoted asymmetric addition of diketene to benzaldehyde.(R)-5-Hydroxy-3-oxo-5-phenyl-pentanoic acid isopropyl esterC14H18O4Er = 85:15[α]D23=+42.4 (c 0.66, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (R)
Co-reporter:Rosa M Moreno, Malgorzata Rosol, Albert Moyano
Tetrahedron: Asymmetry 2006 Volume 17(Issue 7) pp:1089-1103
Publication Date(Web):3 April 2006
DOI:10.1016/j.tetasy.2006.03.016
The condensation of a set of diversely substituted (S)-2-amino-2-ferrocenyl ethanol derivatives 1a–e with the salicylaldehydes 5A–C resulted in the generation of a small library of new chiral Schiff base-ligands, whose titanium isopropoxide complexes have been tested as catalysts in the asymmetric addition of trimethylsilyl cyanide to aldehydes. The enantioselectivity of the reaction is strongly influenced (a) by the substitution pattern of the 2-amino-2-ferrocenyl ethanol moiety, and (b) by the nature of the C3′ substituent in the salicylaldehyde. Computational modelling of the intermediate transition state complexes (based on the Schiff base, benzaldehyde, isopropoxide and cyanide bonded to titanium) show that the experimental results can be accommodated by a careful analysis of the steric interactions between the 2-aminoethanol and salicylaldehyde moieties, and the metal-coordinated aldehyde.Chiral Schiff base–alkoxytitanium complexes derived from salicylaldehydes and from a set of diversely substituted (S)-2-amino-2-ferrocenylethanols have been prepared and tested as catalysts for the asymmetric addition of trimethylsilyl cyanide to aldehydes.(1S,2R)-1-Azido-1-ferrocenyl-2-propyl acetateC15H17FeN3O2Er = 98:2[α]D23=+91.3 (c 0.560, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (1S,2R)(1S,2R)-1-Amino-1-ferrocenyl-2-propanolC13H17FeNOEr = 98:2[α]D23=+70.1 (c 0.510, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (1S,2R)(1R,2S)-2-Amino-2-ferrocenyl-1-phenylethanolC18H19FeNOEr = 99.5:0.5[α]D23=-11.5 (c 0.260, CH2Cl2)Source of chirality:asymmetric synthesisAbsolute configuration:(1R,2S)2-[((S)-1-Ferrocenyl-2-hydroxyethylimino)methyl]phenolC19H19FeNO2Er = 99:1[α]D23=+203.0 (c 0.280, EtOH)Source of chirality:asymmetric synthesisAbsolute configuration:(S)2-[((1S,2R)-1-Ferrocenyl-2-hydroxypropylimino)methyl]phenolC20H21FeNO2Er = 98:2[α]D23=+481.0 (c 0.570, EtOH)Source of chirality:asymmetric synthesisAbsolute configuration:(1S,2R)2-[((S)-1-Ferrocenyl-2-hydroxy-2-methylpropylimino)methyl]phenolC21H23FeNO2Er = 97:3[α]D23=+443.0 (c 0.290, EtOH)Source of chirality:asymmetric synthesisAbsolute configuration:(S)2-tert-Butyl-6-[((S)-1-ferrocenyl-2-hydroxyethylimino)methyl]phenolC23H27FeNO2Er = 99:1[α]D23=+240.0 (c 0.290, EtOH)Source of chirality:asymmetric synthesisAbsolute configuration:(S)2-tert-Butyl-6-[((1S,2R)-1-ferrocenyl-2-hydroxypropylimino)methyl]phenolC24H29FeNO2Er = 98:2[α]D23=+469.0 (c 0.210, EtOH)Source of chirality:asymmetric synthesisAbsolute configuration:(1S,2R)2-tert-Butyl-6-[((S)-1-ferrocenyl-2-hydroxy-2-methylpropylimino)methyl]phenolC24H29FeNO2Er = 97:3[α]D23=+419.2 (c 0.230, EtOH)Source of chirality:asymmetric synthesisAbsolute configuration:(S)2-tert-Butyl-6-[((S)-1-ferrocenyl-2-hydroxy-1-methylethylimino)methyl]phenolC24H29FeNO2Er = 96:4[α]D23=+139.2 (c 0.80, CHCl3)Source of chirality:asymmetric synthesisAbsolute configuration:(S)2-Adamantyl-5-methyl-6-[((S)-1-ferrocenyl-2-hydroxyethylimino)methyl]phenolC30H35FeNO2Er = 99:1[α]D23=+204.2 (c 0.360, EtOH)Source of chirality:asymmetric synthesisAbsolute configuration:(S)2-Adamantyl-5-methyl-6-[((1S,2R)-1-ferrocenyl-2-hydroxypropylimino)methyl]phenolC31H37FeNO2Er = 98:2[α]D23=+408.3 (c 0.570, EtOH)Source of chirality:asymmetric synthesisAbsolute configuration:(1S,2R)2-Adamantyl-5-methyl-6-[((S)-1-ferrocenyl-2-hydroxy-1-methylethylimino)methyl]phenolC31H37FeNO2Er = 96:4[α]D23=+137.6 (c 0.50, EtOH)Source of chirality: asymmetric synthesisAbsolute configuration: (S)2-[((S)-1-Ferrocenyl-2-hydroxy-2-methylpropylamino)methyl]phenolC21H25FeNO2Er = 97:3[α]D23=+61.9 (c 1.350, EtOH)Source of chirality: asymmetric synthesisAbsolute configuration: (S)2-tert-Butyl-6-[((S)-1-ferrocenyl-2-hydroxyethylamino)methyl]phenolC23H29FeNO2Er = 99:1[α]D23=+13.4 (c 0.400, EtOH)Source of chirality: asymmetric synthesisAbsolute configuration: (S)2-tert-Butyl-6-[((1S,2R)-1-ferrocenyl-2-hydroxypropylamino)methyl]phenolC24H31FeNO2Er = 98:2[α]D23=+180.8 (c 0.520, EtOH)Source of chirality: asymmetric synthesisAbsolute configuration: (1S,2R)
Co-reporter:Malgorzata Rosol, Albert Moyano
Journal of Organometallic Chemistry 2005 Volume 690(Issue 9) pp:2291-2296
Publication Date(Web):29 April 2005
DOI:10.1016/j.jorganchem.2005.02.035
1′-Carbopalladated complexes derived from 4-ferrocenyl-1,3-oxazolines are reported in this paper as efficient catalysts for the Heck coupling of iodo- and bromoarenes with alkenes. Experimental evidence points out strongly towards the involvement of a Pd(0)/Pd(II) catalytic cycle in the mechanism of the reaction. For the first time, the disassembly of the carbopalladated complex via coupling with the olefin in a non-catalytic Heck reaction has been demonstrated to be the origin of the release of Pd(0) from the palladacycle carrier.1′-Carbopalladated complexes derived from 4-ferrocenyl-1,3-oxazolines behave as pre-catalysts for the Heck coupling of iodo- and bromoarenes with alkenes. The reaction proceeds through a Pd(0)/Pd(II) catalytic cycle. The release of Pd(0) from the palladacycle takes place by coupling with the olefin in a non-catalytic Heck reaction.
Co-reporter:Ramon Rios, Sergi Paredes, Miquel A. Pericàs, Albert Moyano
Journal of Organometallic Chemistry 2005 Volume 690(Issue 2) pp:358-362
Publication Date(Web):17 January 2005
DOI:10.1016/j.jorganchem.2004.09.046
When the dicobalt(hexacarbonyl) complex of N-(2-butynoyl)-4,4-dimethyloxazolidinone (1) is treated with chiral cyclopentadienyl (tricarbonyl)molybdenum anions, pairs of diastereomeric heterobimetallic (Co–Mo) complexes are obtained. In one instance, the two diastereomers have been separated by chromatography and they have been reacted with norbornadiene; each diastereomer leads with virtually complete stereocontrol to a single enantiomer of the endo Pauson–Khand cycloadduct 5.When the dicobalt(hexacarbonyl) complex of N-(2-butynoyl)-4,4-dimethyloxazolidinone (1) is treated with chiral cyclopentadienyl (tricarbonyl)molybdenum anions, pairs of diastereomeric heterobimetallic (Co–Mo) complexes are obtained. In one instance, the two diastereomers have been separated by chromatography and they have been reacted with norbornadiene; each diastereomer leads with virtually complete stereocontrol to a single enantiomer of the endo Pauson–Khand cycloadduct 5.
Co-reporter:Agustí Bueno, Rosa M Moreno, Albert Moyano
Tetrahedron: Asymmetry 2005 Volume 16(Issue 10) pp:1763-1778
Publication Date(Web):23 May 2005
DOI:10.1016/j.tetasy.2005.03.031
Evidence gathered both from X-ray diffraction data and from molecular modelling studies suggests that the introduction of a gem-dimethyl moiety at C1 in 2-amino-2-ferrocenylethanol should exert strong conformational control on the ferrocenyl group when this compound is incorporated into a heterocyclic ring. In order to test this hypothesis, we have developed an efficient, enantioselective and enantiodivergent route to 1-amino-1-ferrocenyl-2-methylpropan-2-ol. According to our expectations, the corresponding 1,3-oxazolidin-2-one (Fc-‘SuperQuat’) exhibited excellent diastereofacial selectivity in the Diels–Alder reaction of its N-crotonyl derivative with cyclopentadiene, in sharp contrast to the lack of diastereoselectivity observed when using the unsubstituted 4-ferrocenyl-1,3-oxazolidin-2-one. In a similar way, the presence of a gem-dimethyl group at C5 in a 2-(2-diphenylphosphinophenyl)-4-ferrocenyl-1,3-oxazoline (Fc-PHOX) ligand brings about a major change in the enantioselectivity of the palladium-catalyzed asymmetric allylic substitution by dimethyl malonate anion, and leads to unprecedented (for PHOX ligands derived from acyclic 2-amino alcohols) levels of asymmetric induction in substrates such as (E)-1,3-dimethyl-2-propenyl and 2-cyclohexenyl acetates.The introduction of a gem-dimethyl moiety at C1 in 2-amino-2-ferrocenylethanol exerts a strong conformational control on the ferrocenyl group when this compound is incorporated into a heterocyclic ring, leading to increased levels of asymmetric induction in reactions mediated by chiral auxiliaries or ligands derived from this chiral β-amino alcohol.(R)-1-Ferrocenyl-2-methylpropane-1,2-diolC14H18FeO2Ee = 96%[α]D23=-102 (c 1.15, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (R)(S)-1-Ferrocenyl-2-methylpropane-1,2-diolC14H18FeO2Ee = 94%[α]D23=+97.5 (c 1.135, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (S)(R)-1-Amino-1-ferrocenyl-2-methylpropan-2-olC14H19FeNO2Ee = 96%[α]D23=-83.4 (c 0.356, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (R)(S)-1-Amino-1-ferrocenyl-2-methylpropan-2-olC14H19FeNO2Ee = 94%[α]D23=+71.5 (c 0.397, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (S)1,1′-Bis[(S,S)-1,2-dihydroxy-2-methylpropyl]ferroceneC18H26FeO4Ee = 99.2%[α]D23=+33.5 (c 0.097, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (S,S)1,1′-Bis[(S,S)-1-azido-2-hydroxy-2-methylpropyl]ferroceneC18H24FeN6O2Ee = 99.2%[α]D23=+99.3 (c 0.210, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (S,S)(R)-4-Ferrocenyl-5,5-dimethyl-1,3-oxazolidin-2-oneC15H17FeNO2Ee = 96%[α]D23=-194.3 (c 0.356, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (R)(R)-4-Ferrocenyl-5,5-dimethyl-N-(2-(E)-propenoyl)-1,3-oxazolidin-2-oneC19H21FeNO3Ee = 96%[α]D23=+45.5 (c 0.150, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (R)(4R)-3-[(3′S,4′S,5′R,6′R)-5′-Methylbicyclo[2.2.1]hepten-4′-carbonyl]-5,5-dimethyl-4-ferrocenyl-1,3-oxazolidin-2-oneC24H27FeNO3Ee = 96%[α]D23=+64.9 (c 0.160, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration:(4R,3′S,4′S,5′R,6′R)(4R)-3-[(3′S,4′R,5′S,6′R)-5′-Methylbicyclo[2.2.1]hepten-4′-carbonyl]-5,5-dimethyl-4-ferrocenyl-1,3-oxazolidin-2-oneC24H27FeNO3Ee = 96%[α]D23=+33.7 (c 0.170, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration:(4R,3′R,4′S,5′S,6′R)(S)-2-Fluoro-N-(2-hydroxy-1-ferrocenyl-2-methylpropyl)benzamideC21H22FFeNO2Ee = 94%[α]D23=+53.3 (c 0.250, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (S)(S)-2-(2-Fluorophenyl)-4-ferrocenyl-5,5-dimethyl-1,3-oxazolineC21H20FFeNOEe = 94%[α]D23=+364 (c 0.335, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (S)(S)-2-(2-(Diphenylphosphanyl)phenyl)-4-ferrocenyl-5,5-dimethyl-1,3-oxazolineC33H31FeNOPEe = 94%[α]D23=+203 (c 0.220, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (S)[(S)-Diphenyl-(2′-(4-ferrocenyl-5,5-dimethyl(1,3-oxazolin-2-yl)phenyl))phosphine]-[π-allyl]palladium(II) hexafluorophosphateC36H35F6FeNOP2PdEe = 94%[α]D23=-161 (c 0.52, EtOH)Source of chirality: asymmetric synthesisAbsolute configuration: (S)
Co-reporter:Albert Moyano ;Malgorzata Rosol;Rosa M. Moreno;Concepción López ;Miguel A. Maestro
Angewandte Chemie 2005 Volume 117(Issue 12) pp:
Publication Date(Web):18 FEB 2005
DOI:10.1002/ange.200462434
Eine Kohlenstoff-Palladium-Verknüpfung mit dem unsubstituierten Cyclopentadienylring führt im Zuge der Cyclopalladierung von 4-Ferrocenyl-1,3-oxazolinen zu neuartigen metallierten Ferrocenen (siehe Schema). Diese chiralen Komplexe katalysieren die asymmetrische Aza-Claisen-Umlagerung von Allylimidaten.
Co-reporter:Albert Moyano ;Malgorzata Rosol;Rosa M. Moreno;Concepción López ;Miguel A. Maestro
Angewandte Chemie International Edition 2005 Volume 44(Issue 12) pp:
Publication Date(Web):18 FEB 2005
DOI:10.1002/anie.200462434
Formation of a carbon–palladium bond to the unsubstituted cyclopentadiene ring occurs during the cyclopalladation of 4-ferrocenyl-1,3-oxazolines and leads to a new type of metalated ferrocene (see scheme). These chiral complexes catalyze the asymmetric aza-Claisen rearrangement of allylic imidates.
Co-reporter:Xavier Companyó, Guillem Valero, Victor Ceban, Teresa Calvet, Mercé Font-Bardía, Albert Moyano and Ramon Rios
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 23) pp:NaN7989-7989
Publication Date(Web):2011/09/13
DOI:10.1039/C1OB06308A
The highly enantioselective asymmetric allylic alkylation of Morita–Baylis–Hillman carbonates with bis(phenylsulfonyl)methane is presented. The reaction is simply catalyzed by cinchona alkaloid derivatives affording the final alkylated products in good yields and enantioselectivities.
Co-reporter:Xavier Companyó, Guillem Valero, Oriol Pineda, Teresa Calvet, Mercè Font-Bardía, Albert Moyano and Ramon Rios
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 2) pp:NaN439-439
Publication Date(Web):2011/10/06
DOI:10.1039/C1OB06503C
An enantioselective α-oxyamination of unprotected 3-substituted oxindoles with nitrosobenzene catalyzed by tertiary amine–thiourea bifunctional organocatalysts has been developed and affords the corresponding 3-amino-2-oxindole derivatives in good yields and with moderate to excellent enantioselectivities (up to > 99.9:0.1 er when the product is isolated by direct filtration from the reaction mixture). The absolute configuration of the major enantiomers of the products has been established both by chemical correlation and by comparison between the theoretically calculated and the experimental ECD.

![Benzenepropanoic acid, 2-bromo-β-[[(1,1-dimethylethoxy)carbonyl]oxy]-α-methylene-, methyl ester](/data/chemimg/141200/1202888-62-7.png)
![Benzenepropanoic acid, 2-bromo-β-[[(1,1-dimethylethoxy)carbonyl]oxy]-α-methylene-, methyl ester](/data/chemimg/141200/1202888-62-7_b.png)
![Benzenepropanoic acid, β-[[(1,1-dimethylethoxy)carbonyl]oxy]-4-methyl-α-methylene-, methyl ester](/data/chemimg/167300/1187847-13-7.png)
![Benzenepropanoic acid, β-[[(1,1-dimethylethoxy)carbonyl]oxy]-4-methyl-α-methylene-, methyl ester](/data/chemimg/167300/1187847-13-7_b.png)
![Benzenepropanoic acid, β-[[(1,1-dimethylethoxy)carbonyl]oxy]-4-methoxy-α-methylene-, methyl ester](/data/chemimg/141200/1132641-34-9.png)
![Benzenepropanoic acid, β-[[(1,1-dimethylethoxy)carbonyl]oxy]-4-methoxy-α-methylene-, methyl ester](/data/chemimg/141200/1132641-34-9_b.png)
![Benzenepropanoic acid, β-[[(1,1-dimethylethoxy)carbonyl]oxy]-4-fluoro-α-methylene-, methyl ester](/data/chemimg/141200/1132641-31-6.png)
![Benzenepropanoic acid, β-[[(1,1-dimethylethoxy)carbonyl]oxy]-4-fluoro-α-methylene-, methyl ester](/data/chemimg/141200/1132641-31-6_b.png)
![Benzenepropanoic acid, 4-chloro-β-[[(1,1-dimethylethoxy)carbonyl]oxy]-α-methylene-, methyl ester](/data/chemimg/141200/1132641-29-2.png)
![Benzenepropanoic acid, 4-chloro-β-[[(1,1-dimethylethoxy)carbonyl]oxy]-α-methylene-, methyl ester](/data/chemimg/141200/1132641-29-2_b.png)
![1-Naphthalenepropanoic acid, β-[[(1,1-dimethylethoxy)carbonyl]oxy]-α-methylene-, methyl ester](/data/chemimg/141200/1005193-01-0.png)
![1-Naphthalenepropanoic acid, β-[[(1,1-dimethylethoxy)carbonyl]oxy]-α-methylene-, methyl ester](/data/chemimg/141200/1005193-01-0_b.png)
![Benzenepropanoic acid, β-[[(1,1-dimethylethoxy)carbonyl]oxy]-α-methylene-, methyl ester](/data/chemimg/105500/956833-12-8.png)
![Benzenepropanoic acid, β-[[(1,1-dimethylethoxy)carbonyl]oxy]-α-methylene-, methyl ester](/data/chemimg/105500/956833-12-8_b.png)
![2-Butanone, 4-(4-chlorophenyl)-3-hydroxy-4-[(4-methoxyphenyl)amino]-,(3R,4R)-](http://img.cochemist.com/ccimg/925500/925454-93-9.png)
![2-Butanone, 4-(4-chlorophenyl)-3-hydroxy-4-[(4-methoxyphenyl)amino]-,(3R,4R)-](http://img.cochemist.com/ccimg/925500/925454-93-9_b.png)







![Acetic acid, [(4-methoxyphenyl)imino]-, ethyl ester, (2E)-](http://img.cochemist.com/ccimg/120500/120419-96-7.png)
![Acetic acid, [(4-methoxyphenyl)imino]-, ethyl ester, (2E)-](http://img.cochemist.com/ccimg/120500/120419-96-7_b.png)