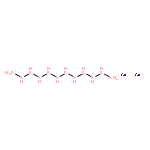
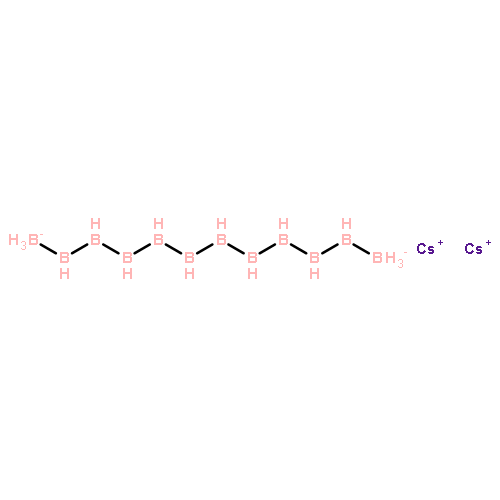
Co-reporter:Zhao Li, Teng He, Daiju Matsumura, Shu Miao, Anan Wu, Lin Liu, Guotao Wu, and Ping Chen
ACS Catalysis October 6, 2017 Volume 7(Issue 10) pp:6762-6762
Publication Date(Web):August 21, 2017
DOI:10.1021/acscatal.7b01790
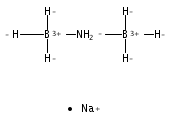
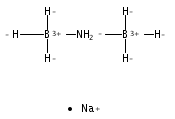
The development of cost-effective and highly efficient catalysts is of scientific importance and practical need in the conversion and utilization of clean energy. One of the strategies fulfilling that demand is to achieve high exposure of a catalytically functional noble metal to reactants to maximize its utilization efficiency. We report herein that the single-atom alloy (SAA) made of atomically dispersed Pt on the surface of Ni particles (Pt is surrounded by Ni atoms) exhibits improved catalytic activity on the hydrolytic dehydrogenation of ammonia–borane, a promising hydrogen storage method for onboard applications. Specifically, an addition of 160 ppm of Pt leads to ca. 3-fold activity improvement in comparison to that of pristine Ni/CNT catalyst. The turnover frequency based on the isolated Pt is 12000 molH2 molPt–1 min–1, which is about 21 times the value of the best Pt-based catalyst ever reported. Our simulation results indicate that the high activity achieved stems from the synergistic effect between Pt and Ni, where the negatively charged Pt (Ptδ-) and positively charged Ni (Niδ+) in the Pt-Ni alloy are prone to interact with H and OH of H2O molecules, respectively, leading to an energetically favorable reaction pathway.Keywords: ammonia borane; catalytic dehydrogenation; hydrogen storage; Pt-Ni alloy; single-atom alloy;
Co-reporter:Zhao Li;Lin Liu;Weidong Chen;Miao Zhang;Guotao Wu;Ping Chen
Chemical Science (2010-Present) 2017 vol. 8(Issue 1) pp:781-788
Publication Date(Web):2016/12/19
DOI:10.1039/C6SC02456D
Development of non-noble metal catalysts with similar activity and stability to noble metals is of significant importance in the conversion and utilization of clean energy. The catalytic hydrolysis of ammonia borane (AB) to produce 3 equiv. of H2, as an example of where noble metal catalysts significantly outperform their non-noble peers, serves as an excellent test site for the design and optimization of non-noble metal catalysts. Our kinetic isotopic effect measurements reveal, for the first time, that the kinetic key step of the hydrolysis is the activation of H2O. Deducibly, a transition metal with an optimal electronic structure that bonds H2O and –OH in intermediate strengths would favor the hydrolysis of AB. By employing a covalent triazine framework (CTF), a newly developed porous material capable of donating electrons through the lone pairs on N, the electron densities of nano-sized Co and Ni supported on CTF are markedly increased, as well as their catalytic activities. Specifically, Co/CTF exhibits a total turnover frequency of 42.3 molH2 molCo−1 min−1 at room temperature, which is superior to all peer non-noble metal catalysts ever reported and even comparable to some noble metal catalysts.
Co-reporter:Miao Zhang;Qijun Pei;Weidong Chen;Lin Liu;Ping Chen
RSC Advances (2011-Present) 2017 vol. 7(Issue 8) pp:4306-4311
Publication Date(Web):2017/01/10
DOI:10.1039/C6RA26667C
Reduced TiO2 (TiO2−x) materials have attracted increasing attention due to their large solar absorption and high photo-activity. However, their synthesis procedures usually involve harsh conditions, such as high temperature and/or high pressure. Herein, a facile solid ball-milling method for the synthesis of TiO2−x under ambient conditions was developed. By using finely dispersed Na/NaCl powders as the reducing agent and TiO2 (P25, Degussa) as the precursor, a series of TiO2−x of 20–30 nm with a controllable reduction degree can be successfully synthesized through adjusting the reaction conditions. The surface area of TiO2−x is much larger than that of pristine TiO2, showing its great potential as a catalyst support in chemical reactions. Our experimental results show that uniform Ru particles with particle size less than 1 nm can be well dispersed on the surface of the TiO2−x due to the enhanced surface area and plenty of oxygen vacancies in TiO2−x. As a result, Ru/TiO2−x exhibited superior activity upon catalytic hydrogenation of N-methylpyrrole in comparison with Ru/TiO2.
Co-reporter:Zhao Li, Teng He, Guotao Wu, Weidong Chen, Yong Shen Chua, Jianping Guo, Dong Xie, Xiaohua Ju and Ping Chen
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 1) pp:244-251
Publication Date(Web):05 Nov 2015
DOI:10.1039/C5CP04257G
The calcium amidoborane hydrazinates, Ca(NH2BH3)2·nN2H4, were firstly synthesized by reacting different molar ratios of Ca(NH2BH3)2 and N2H4. In particular, Ca(NH2BH3)2 and N2H4 with a molar ratio of 1:2 crystallizes into the orthorhombic symmetry P212121 space group with the lattice parameters of a = 6.6239(4) Å, b = 13.7932(6) Å, c = 4.7909(2) Å. The dehydrogenations of calcium amidoborane hydrazinates are two-step reactions, exhibiting superior dehydrogenation properties compared with those of pristine Ca(NH2BH3)2. For Ca(NH2BH3)2–1/2N2H4, approximately 4.6 equiv. hydrogen (or 7.9 wt% hydrogen) can be released at 150 °C. Kinetic analysis shows that the activation energies for the two steps of hydrogen desorption from Ca(NH2BH3)2·2N2H4 are much lower than those of pristine Ca(NH2BH3)2, suggesting an improvement in the dehydrogenation kinetics of Ca(NH2BH3)2 after coordinating with N2H4. Isotopic labeling results show that the driving force for the dehydrogenation of calcium amidoborane hydrazinates is the combination mechanism of protonic hydrogen and hydridic hydrogen (Hδ+ and Hδ−). In addition, initial H2 release from calcium amidoborane hydrazinates originates from the interaction of [–BH3] and N2H4, rather than [–BH3] and [–NH2] (in [–NH2BH3]).
Co-reporter:Teng He, Lin Liu, Guotao Wu and Ping Chen
Journal of Materials Chemistry A 2015 vol. 3(Issue 31) pp:16235-16241
Publication Date(Web):03 Jul 2015
DOI:10.1039/C5TA03056K
A covalent triazine framework (CTF) with high surface area, large amount of nitrogen functionalities, and high porosity and basicity was employed as a support for palladium nanoparticles (NPs). A well-dispersed Pd/CTF-1 catalyst with uniform distribution of Pd particles was successfully synthesized in the present study. The as-prepared 4% Pd/CTF-1 catalyst showed a markedly improved activity in the hydrogenation of N-heterocyclic compounds compared to the activated carbon (AC)-supported catalyst, i.e., the Pd/CTF-1 catalyst exhibits ca. 3.6 times faster reaction than Pd/AC in the hydrogenation of N-methylpyrrole. Characterization of Pd/CTF indicated electron donation from the N in CTF to the metallic Pd NPs, showing intensified electronic interaction between the Pd NPs and CTF support, which is responsible for the enhanced activities for the catalytic hydrogenation of N-heterocycles.
Co-reporter:Zhao Li, Teng He, Guotao Wu, Xiaohua Ju, Ping Chen
International Journal of Hydrogen Energy 2015 Volume 40(Issue 15) pp:5333-5339
Publication Date(Web):27 April 2015
DOI:10.1016/j.ijhydene.2015.01.114
Co-reporter:Hujun Cao, Han Wang, Teng He, Guotao Wu, Zhitao Xiong, Jieshan Qiu and Ping Chen
RSC Advances 2014 vol. 4(Issue 61) pp:32555-32561
Publication Date(Web):17 Jul 2014
DOI:10.1039/C4RA02864C
The Mg(NH2)2–2LiH composite is a promising on-board hydrogen storage material due to its high reversible hydrogen capacity and suitable thermodynamic properties. However, the severe kinetic barrier inhibits its low temperature operation. In the present work, the additive effects of lithium halides on the Mg(NH2)2–2LiH system were studied systematically. Experimental results showed that, among all those lithium halides, the LiBr doped Mg(NH2)2–2LiH composite exhibited the best dehydrogenation performance. The hydrogen sorption and desorption rates of the Mg(NH2)2–2LiH–0.2LiBr sample are ∼3 and 2 times, respectively, faster than that of the pristine sample at 140 °C. At the same time, enhanced kinetics for hydrogen desorption was observed from an activation energy (Ea) of ca. 92 ± 9 kJ mol−1 which was significantly decreased by 35 kJ mol−1 compared with the pristine sample. Subsequently, a plausible mechanism for the modified dehydrogenation/re-hydrogenation process was proposed.
Co-reporter:Teng He, Qijun Pei, Ping Chen
Journal of Energy Chemistry (September 2015) Volume 24(Issue 5) pp:587-594
Publication Date(Web):1 September 2015
DOI:10.1016/j.jechem.2015.08.007
The development of efficient hydrogen storage materials is one of the biggest technical challenges for the coming “hydrogen economy”. The liquid organic hydrogen carriers (LOHCs) with high hydrogen contents, reversibilities and moderate dehydrogenation kinetics have been considered as an alternative option supplementing the extensively investigated inorganic hydride systems. In this review, LOHCs for long distance H2 transport and for onboard application will be discussed with the focuses of the design and development of LOHCs and their hydrogenation & dehydrogenation catalyses.liquid organic hydrogen carriers (LOHCs) with high hydrogen content, moderate operational temperature, and the compatibility with existing gasoline infrastructure, hold the promises as hydrogen carriers for both onboard application and large scale long-distance H2 transportation.Download high-res image (165KB)Download full-size image
Co-reporter:Zhao Li, Teng He, Lin Liu, Weidong Chen, Miao Zhang, Guotao Wu and Ping Chen
Chemical Science (2010-Present) 2017 - vol. 8(Issue 1) pp:NaN788-788
Publication Date(Web):2016/08/30
DOI:10.1039/C6SC02456D
Development of non-noble metal catalysts with similar activity and stability to noble metals is of significant importance in the conversion and utilization of clean energy. The catalytic hydrolysis of ammonia borane (AB) to produce 3 equiv. of H2, as an example of where noble metal catalysts significantly outperform their non-noble peers, serves as an excellent test site for the design and optimization of non-noble metal catalysts. Our kinetic isotopic effect measurements reveal, for the first time, that the kinetic key step of the hydrolysis is the activation of H2O. Deducibly, a transition metal with an optimal electronic structure that bonds H2O and –OH in intermediate strengths would favor the hydrolysis of AB. By employing a covalent triazine framework (CTF), a newly developed porous material capable of donating electrons through the lone pairs on N, the electron densities of nano-sized Co and Ni supported on CTF are markedly increased, as well as their catalytic activities. Specifically, Co/CTF exhibits a total turnover frequency of 42.3 molH2 molCo−1 min−1 at room temperature, which is superior to all peer non-noble metal catalysts ever reported and even comparable to some noble metal catalysts.
Co-reporter:Zhao Li, Teng He, Guotao Wu, Weidong Chen, Yong Shen Chua, Jianping Guo, Dong Xie, Xiaohua Ju and Ping Chen
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 1) pp:NaN251-251
Publication Date(Web):2015/11/05
DOI:10.1039/C5CP04257G
The calcium amidoborane hydrazinates, Ca(NH2BH3)2·nN2H4, were firstly synthesized by reacting different molar ratios of Ca(NH2BH3)2 and N2H4. In particular, Ca(NH2BH3)2 and N2H4 with a molar ratio of 1:2 crystallizes into the orthorhombic symmetry P212121 space group with the lattice parameters of a = 6.6239(4) Å, b = 13.7932(6) Å, c = 4.7909(2) Å. The dehydrogenations of calcium amidoborane hydrazinates are two-step reactions, exhibiting superior dehydrogenation properties compared with those of pristine Ca(NH2BH3)2. For Ca(NH2BH3)2–1/2N2H4, approximately 4.6 equiv. hydrogen (or 7.9 wt% hydrogen) can be released at 150 °C. Kinetic analysis shows that the activation energies for the two steps of hydrogen desorption from Ca(NH2BH3)2·2N2H4 are much lower than those of pristine Ca(NH2BH3)2, suggesting an improvement in the dehydrogenation kinetics of Ca(NH2BH3)2 after coordinating with N2H4. Isotopic labeling results show that the driving force for the dehydrogenation of calcium amidoborane hydrazinates is the combination mechanism of protonic hydrogen and hydridic hydrogen (Hδ+ and Hδ−). In addition, initial H2 release from calcium amidoborane hydrazinates originates from the interaction of [–BH3] and N2H4, rather than [–BH3] and [–NH2] (in [–NH2BH3]).
Co-reporter:Teng He, Lin Liu, Guotao Wu and Ping Chen
Journal of Materials Chemistry A 2015 - vol. 3(Issue 31) pp:NaN16241-16241
Publication Date(Web):2015/07/03
DOI:10.1039/C5TA03056K
A covalent triazine framework (CTF) with high surface area, large amount of nitrogen functionalities, and high porosity and basicity was employed as a support for palladium nanoparticles (NPs). A well-dispersed Pd/CTF-1 catalyst with uniform distribution of Pd particles was successfully synthesized in the present study. The as-prepared 4% Pd/CTF-1 catalyst showed a markedly improved activity in the hydrogenation of N-heterocyclic compounds compared to the activated carbon (AC)-supported catalyst, i.e., the Pd/CTF-1 catalyst exhibits ca. 3.6 times faster reaction than Pd/AC in the hydrogenation of N-methylpyrrole. Characterization of Pd/CTF indicated electron donation from the N in CTF to the metallic Pd NPs, showing intensified electronic interaction between the Pd NPs and CTF support, which is responsible for the enhanced activities for the catalytic hydrogenation of N-heterocycles.