Co-reporter:Yinglin Shen, Ziyi Liu, Xia Yang, Hui Wang, Zhifang Chai, and Dongqi Wang
Industrial & Engineering Chemistry Research November 8, 2017 Volume 56(Issue 44) pp:12708-12708
Publication Date(Web):October 10, 2017
DOI:10.1021/acs.iecr.7b01742
Solvent extraction studies of UO22+ were carried out using N,N′-dimethyl-N,N′-dibutylmalonamide (DMDBMA), N,N,N′,N′-tetrabutylmalonamide (TBMA), and N,N,N′,N′-tetraoctylmalonamide (TOMA) dissolved in three room temperature ionic liquids (RTILs) of 1-alkyl-3-methylimidazolium bis(trifluoromethane)sulfonamide ([Cnmim][Tf2N], n = 4,6,8). At lower acidity, the extraction of UO22+ carried out in RTIL was found to be much more efficient than in the conventional molecular diluents, while this advantage diminishes at higher acidity. The alkyl chain length of the RTILs plays an important role on the extraction performance. The effect of concentration of three diamides and nitric acid on extraction performance was also investigated. The extraction mechanism using RTILs as the diluent was deduced by the slope analysis and extraction experiment, and a cation-exchange mechanism at lower acidity and a neutral solvation mechanism at higher acidity were proposed. Density functional theory method was used to characterize the key species of the coordination complexes of UO22+ with the three malonamide (MA) ligands in terms of geometry, Mayer bond order (MBO), binding energy, interface exchange reaction, stepwise mechanism, and quantum theory of atom-in-molecule (QTAIM) topological analysis.
Co-reporter:Hongcai Ling;Miaoren Xia;Wenkai Chen;Zhifang Chai
RSC Advances (2011-Present) 2017 vol. 7(Issue 20) pp:12236-12246
Publication Date(Web):2017/02/16
DOI:10.1039/C6RA26114K
The interaction of neptunyl ions (NpO2+) with three picolinic type ligands (L), including the deprotonated picolinic acid anion (PA−), the deprotonated dipicolinic acid anion (DPA2−) and the 1,10-phenanthroline-2,9-dicarboxylic acid anion (PADA2−), was investigated by using a density functional theory method with various stoichiometric ratios of Np : L = 1 : 1, 1 : 2, and 1 : 3. The coordination modes, the influence of the denticity of the ligands, and the stoichiometry of the complexes were evaluated in terms of geometry, electronic structure, and thermodynamics. The calculations show that the coordination of NpO2+ to tetradentate ligands is more stable than that to tridentate and bidentate ones, and the coordination ability of the three deprotonated ligands follows the order: PADA2− > DPA2− > PA−. Quantum theory of atoms-in-molecules (QTAIM) analysis, charge decomposition analysis (CDA) and natural atomic orbital (NAO) analysis were used to understand the bonding nature and electronic properties of the complexes, and the metal–ligand dative bond was identified to be mainly ionic. In view of the favorable coordination modes and the distinct ability of the ligands in binding to neptunyl, we conclude that the denticity of the ligands and the combined hard–soft donor strategy work cooperatively in the coordination of NpO2+ with ligands. This work is expected to contribute to the rational design of new types of ligand with enhanced ability to extract neptunyl.
Co-reporter:Meng Wang;Wanjian Ding
RSC Advances (2011-Present) 2017 vol. 7(Issue 7) pp:3667-3675
Publication Date(Web):2017/01/09
DOI:10.1039/C6RA26109D
Transferrins have been proposed to be responsible for the in vivo transportation of uranyl. In this work, the binding mechanism of uranyl to transferrin has been studied using density functional theory method. Three possible stepwise pathways have been investigated and compared, differing in the sequence of the three residues to bind with uranyl, i.e. Tyr* → Tyr* → Asp* (YYD) and Tyr* → Asp* → Tyr* (YDY) and Asp* → Tyr* → Tyr* (DYY). Compared with the activation energies and the reaction heat of these three possible mechanisms, it is concluded that the YYD pathway is a more plausible description for the binding of uranyl. According to the calculations, the binding process is described as a ligand exchange process assisted by the hydrolysis of uranyl tricarbonate complex, and the role of carbonate ligand which determines the optimal pathway is identified. The QTAIM analysis was used to compare the bond nature of uranyl complexes in its free form and its complex with the amino acid residues. The results are expected to benefit our understanding of the uptake of uranyl by serum transferrins, and have implications on protein engineering and the development of decorporation agents towards improved binding kinetics and thermodynamics of uranyl in a specific pH range.
Co-reporter:Meixiu Yang;Wanjian Ding
New Journal of Chemistry (1998-Present) 2017 vol. 41(Issue 1) pp:63-74
Publication Date(Web):2016/12/19
DOI:10.1039/C6NJ01615D
We reported a density functional theory study on the complexation of six hydrated actinyl cations, AnO2(H2O)52+/+ (aq) (An = U/Np/Pu), with three expanded porphyrins, amethyrin (H4L1), oxasapphyrin (H2L2), and grandephyrin (H3L3). The geometries have been fully optimized and analyzed, and the electronic structures, the binding free energies, and the NMR properties were calculated. Natural population analysis and Quantum Theory of Atoms in Molecules (QTAIM) topology analysis techniques were applied to understand the interaction modes between two entities of each complex. The calculations show that for the same ligand, PuO22+ and NpO22+ display stronger binding affinity than UO22+, UO2+, NpO2+, and PuO2+, and among the three ligands tested, L22− fits better with the actinyl cations than L33− and H2L12−. The redox process was observed in the complexation of PuO22+ and NpO22+ with specific ligands, which agrees with the experimental results. In the characterization of the nature of the coordination bonding interactions, QTAIM gives a consistent description with the natural population analysis method and shows that the interaction between An and the electron donor atoms in the first coordination shell has a strong ionic feature, while the interaction between An and Oyl atoms of the actinyls in the complexes remains covalent. This work complements the earlier experimental studies by providing a molecular level of understanding on the interaction between actinyls and expanded porphyrins, and is expected to contribute to the communities of the chemistry of actinides and expanded porphyrins.
Co-reporter:Chao Zhang, Huai-Zhi Song, Fei Mao, Cheng-Jun Wang, Dong-Qi Wang, Feng-Shou Zhang
Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms 2017 Volume 406, Part B(Volume 406, Part B) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.nimb.2017.04.068
Silicon carbide fiber-reinforced silicon carbide matrix composites have been investigated for their use as structural materials for advanced nuclear reactor. Although quite a number of researches have been devoted to probe the effects of irradiation on various properties of the composites, there is little known about the atomistic mechanism for irradiation resistance. In this study, a two-temperature model has been used to investigate the irradiation damage of SiC/Gra/SiC composites, which includes three parts and two SiC/C interfaces, two single crystal cubic silicon carbide on two sides and a few graphene sheets in the middle part. By simulating 100 keV displacement cascades, we find that the number of defects in the reinforcement is larger than that in the matrix, which indicates the damage in the reinforcement is more serious than that in the matrix. Moreover, we explicitly investigate the damage behavior of the interphase graphene layers and find that some atoms in one graphene sheet form many new chemical bonds with atoms in another one, which leads to the transition from sp2 to sp3 hybridization. The newly formed chemical bonds link the different graphene layers and make graphene-like electronic structure more “diamond-like”, enhancing the irradiation resistance of the matrix.
Co-reporter:Hui Wang, Zhifang Chai, Dongqi Wang
Green Energy & Environment 2017 Volume 2, Issue 1(Volume 2, Issue 1) pp:
Publication Date(Web):1 January 2017
DOI:10.1016/j.gee.2016.11.011
The adsorption of uranyl on hydroxylated α-SiO2(001) in the presence of a series of anionic ligands, i.e. OH−, CO32-, NO3-, H2PO4-, HPO42-, CH3COO− (Ac−), C6H5COO− (PhCO2-), C6H5O− (PhO−), was studied by the periodic density functional theory (DFT) implemented in the Vienna ab initio simulation package (VASP). For the ligands other than OH− and PhO−, only the bidentate coordination modes to the uranyl were considered. The excess charge effect of a charged system was first evaluated by constructing models with net charge as is or neutralized by creating defect at the bottom of silica, and the results show that a neutralized model, even with defects, is more realistic than the charged ones. All uranyl species prefer to bind with the deprotonated site (O−) rather than the protonated one (OH), which suggests that the increase of pH, which leads to the deprotonation of the surface, may enhance the uranyl adsorption. On the other hand, the anionic ligands, which are formed at higher pH, have negative effects. The weaker acidic ligands, such as H2CO3, H3PO4 and H2O, whose speciation in solutions is sensitive to the fluctuation of pH, have more complex effect on the uranyl adsorption than strong acids or bases. Humic substances may coordinate with uranyl through carboxyl and phenolic groups, with the carboxyl group bound stronger. The ternary complexes with one bidentate (or monodentate) anion and one (or two) H2O as ligands, which leads to the uranyl penta-coordinated in its equatorial plane, are more favorable than other configurations when bound to the same anionic ligand. Both the charged nature and the coordination behavior of an anionic ligand are relevant to its ability to influence the adsorption of uranyl on the mineral surface. In addition, the uranyl species adsorbed at the surface functionalized by anionic ligands were also addressed, and the functionalized surfaces have weaker interaction with hydrated uranyl dication.The adsorption of uranyl on hydroxylated α-SiO2[001] in the presence of anions.Download high-res image (190KB)Download full-size image
Co-reporter:Liqun Ye, Hui Wang, Xiaoli Jin, Yurong Su, Dongqi Wang, Haiquan Xie, Xiaodi Liu, Xinxin Liu
Solar Energy Materials and Solar Cells 2016 Volume 144() pp:732-739
Publication Date(Web):January 2016
DOI:10.1016/j.solmat.2015.10.022
•Olive-green few-layered BiOI was successfully synthesized.•Few-layered BiOI displayed expanded facets spacing and oxygen vacancy.•Few-layered BiOI showed higher photo-activity for CO2 conversion than bulk BiOI.Olive-green few-layered BiOI with expanded spacing of the (001) facets and oxygen vacancy is successfully synthesized and characterized. The experimental analysis and theoretical calculation demonstrate that the expanded facets spacing and oxygen vacancy of few-layered BiOI result in enhanced separation efficiency of photoinduced carriers and photon absorption efficiency. Therefore, few-layered BiOI shows much higher photocatalytic activity for CO2 conversion than that of bulk BiOI under visible or near-infrared (NIR) light. And the apparent quantum yields (AQY) are 0.14% and 0.02% for few-layered BiOI under 420 and 700 nm monochromatic light irradiation, respectively. These findings deep our understanding of few-layered BiOX (X=Cl,Br,I) photocatalysts, and propose a new route to design high efficient photocatalysts for energy and environmental photocatalysis.Olive-green few-layered BiOI with expanded spacing of the (001) facets and oxygen vacancy was synthesized for efficient photocatalytic CO2 conversion under visible/near-infrared light.Download high-res image (233KB)Download full-size image
Co-reporter:Tu Lan, Hui Wang, Jiali Liao, Yuanyou Yang, Zhifang Chai, Ning Liu, and Dongqi Wang
Environmental Science & Technology 2016 Volume 50(Issue 20) pp:11121-11128
Publication Date(Web):September 26, 2016
DOI:10.1021/acs.est.6b03583
This work targeted a molecular level of understanding on the dynamics of humic acid (HA) and its interaction with uranyl in the presence of hydrophobic surface mimicked by a carbon nanotube (CNT), which also represents a potential intruder in the environment accompanying with the development of nanotechnology. In aqueous phase, uranyl and HA were observed to build close contact spontaneously, driven by electrostatic interaction, leading to a more compact conformation of HA. The presence of CNT unfolds HA via π–π interactions with the aromatic rings of HA without significant perturbation on the interaction strength between HA and uranyl. These results show that the hydrophilic uranyl and the hydrophobic CNT influence the folding behavior of HA in distinct manners, which represents two fundamental mechanisms that the folding behavior of HA may be modulated in the environment, that is, uranyl enhances the folding of HA via electrostatic interactions, whereas CNT impedes its spontaneous folding via van der Waals (vdW) interactions. The work also provides molecular level of evidence on the transformation of a hydrophobic surface into a hydrophilic one via noncovalent functionalization by HA, which in turn affects the migration of HA and the cations it binds to.
Co-reporter:Chao Zhang, Fei Mao, Xiang-Rui Meng, Dong-Qi Wang, Feng-Shou Zhang
Chemical Physics Letters 2016 Volume 657() pp:184-189
Publication Date(Web):16 July 2016
DOI:10.1016/j.cplett.2016.06.009
Highlights
- •
The coalescence processes of two single-walled carbon nanotubes are studied by coaxial collision.
- •
Five impact cases are investigated to explore the coalescence processes of the nanotubes.
- •
Graphene sheets, graphene nanoribbons, and single-walled CNTs are created after collision.
- •
Some defects formed in the carbon nanomaterials can be excluded by quenching and annealing.
Co-reporter:Xiaoyan Zhou, Bing Wang, Tu Lan, Hanqing Chen, Hailong Wang, Ye Tao, Zhihong Li, Kurash Ibrahim, Dongqi Wang, and Weiyue Feng
The Journal of Physical Chemistry C 2016 Volume 120(Issue 45) pp:25789-25795
Publication Date(Web):November 2, 2016
DOI:10.1021/acs.jpcc.6b07582
Graphene oxide (GO) with an extraordinary atomic and electronic structure has germinated into an attractive building block for the design and fabrication of new chiral nanomaterials. Herein, we have synthesized a GO–humic acid (HA) sandwich-type complex that can self-assemble into a twisted, long-range-ordered nanostructure in aqueous media. The GO–HA sandwich complex shows obvious chirality under circular dichroism (CD) measurement. By means of scanning electron microscopy, small-angle X-ray scattering, confocal microscopy, and polarized optical microscopy (POM), the morphology, size, fine texture, and orientation of the assembly nanostructure of GO–HA have been validated. The results of experiments and molecular dynamics simulations reveal that the interactions between GO and HA (via electrostatic repulsive, π–π stacking, hydrogen bond, and solvation) induce the enhanced formation of natural ripples in a single GO–HA sheet, cause interlayer and interdomain distortion, and generate a twisted conformation of the C═C double bond, which contributes to the induction of the macroscopic chirality of the GO–HA complex.
Co-reporter:Bing Wang, Xiaoyan Zhou, Dongqi Wang, Jun-Jie Yin, Hanqing Chen, Xingfa Gao, Jing Zhang, Kurash Ibrahim, Zhifang Chai, Weiyue Feng and Yuliang Zhao
Nanoscale 2015 vol. 7(Issue 6) pp:2651-2658
Publication Date(Web):23 Dec 2014
DOI:10.1039/C4NR06665K
Preparation of heterogeneous catalysts with active ferrous centers is of great significance for industrial and environmental catalytic processes. Nanostructured carbon materials (NCM), which possess free-flowing π electrons, can coordinate with transition metals, provide a confinement environment for catalysis, and act as potential supports or ligands to construct analogous complexes. However, designing such catalysts using NCM is still seldom studied to date. Herein, we synthesized a sandwich structured ternary complex via the coordination of Fe-loaded humic acid (HA) with CC bonds in the aromatic rings of carbon nanotubes (CNTs), in which the O/N–Fe–C interface configuration provides the confinement environment for the ferrous sites. The experimental and theoretical results revealed octahedrally/tetrahedrally coordinated geometry at Fe centers, and the strong hybridization between CNT C π* and Fe 3d orbitals induces discretization of the atomic charges on aromatic rings of CNTs, which facilitates O2 adsorption and electron transfer from carbon to O2, which enhances O2 activation. The O2 activation by the novel HA/Fe-CNT complex can be applied in the oxidative degradation of phenol red (PR) and bisphenol A (BPA) in aqueous media.
Co-reporter:Hui Wang, Zhifang Chai and Dongqi Wang
Dalton Transactions 2015 vol. 44(Issue 4) pp:1646-1654
Publication Date(Web):12 Nov 2014
DOI:10.1039/C4DT02872D
The adsorption of [UO2(H2O)5]2+ on a hydroxylated α-SiO2(001) surface was studied by periodic density functional theory (DFT) and ab initio molecular dynamics (AIMD) simulation. The effects of pH, CO2, aqua solution and anionic ligands (OH−, NO3− and Cl−) on the adsorption geometry and stability were investigated. The results show that the adsorption of uranyl on a hydroxylated α-SiO2(001) surface leads to the formation of inner-sphere complexes, in which the bidentate complex at the double deprotonated site is most favored. The binding strengths of bidentate and monodentate complexes at the same site are similar, and they become weaker as the number of protons increases at the adsorption site, indicating an enhancement of the adsorption strength at higher pH values within a certain range. Strong chemical interaction plays an important role in all inner-sphere complexes. The hydrogen bonds are formed between uranyl and the hydroxylated surface in all inner- and outer-sphere complexes. The presence of CO2 weakens the adsorption of uranyl on the surface by forming uranyl carbonate (CO32−, HCO3−) complexes. The effect of the anion ligands depends on their charged state and their concentration in solutions. The explicit treatment of water environment in the models has a slight effect on the adsorption configuration. These results are consistent with the experimental observations.
Co-reporter:Xia Yang, Zhifang Chai and Dongqi Wang
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 11) pp:7537-7547
Publication Date(Web):12 Feb 2015
DOI:10.1039/C4CP04586F
Four types of reaction mechanisms for the oxo ligand exchange of monomeric and dimeric neptunyl(VI) hydroxide in aqueous solution were explored computationally using density functional theory (DFT) and ab initio classical molecular dynamics. The obtained results were compared with previous studies on the oxo exchange of uranyl hydroxide, as well as with experiments. It is found that the stable T-shaped [NpO3(OH)3]3− intermediate is a key species for oxo exchange in the proton transfer in mononuclear Path I and binuclear Path IV, similar to the case of uranyl(VI) hydroxide. Path I is thought to be the preferred oxo exchange mechanism for neptunyl(VI) hydroxide in our calculations, due to the lower activation energy (22.7 and 13.1 kcal mol−1 for ΔG‡ and ΔH‡, respectively) of the overall reaction. Path II via a cis-neptunyl structure assisted by a water molecule might be a competitive channel against Path I with a mononuclear mechanism, owing to a rapid dynamical process occurring in Path II. In Path IV with the binuclear mechanism, oxo exchange is accomplished via the interaction between [NpO2(OH)4]2− and T-shaped [NpO3(OH)3]3− with a low activation energy for the rate-determining step, however, the overall energy required to fulfill the reaction is slightly higher than that in mononuclear Path I, suggesting a possible binuclear process in the higher energy region. The chemical bonding evolution along the reaction pathways was discussed by using topological methodologies of the electron localization function (ELF).
Co-reporter:Hui Wang, Jing-Yao Liu, Zhifang Chai and Dongqi Wang
RSC Advances 2015 vol. 5(Issue 7) pp:4909-4917
Publication Date(Web):10 Dec 2014
DOI:10.1039/C4RA15368E
The activation of CO, hydrogenation of CHx (x = 0–4) and C2Hy (y = 0–5) species and carbon chain propagation on V(100) were studied by means of periodic density functional theory (DFT) calculations. The results indicate that the activation of CO is very facile on V(100) via direct dissociation rather than H-assisted pathways. The hydrogenation of CHx/C2Hy (except for CC) and the C–C coupling elementary steps are thermodynamically and kinetically unfavorable. The energy barriers to the former reactions are lower than those to the latter ones. The high coverage of reactants and the entropic effect may be the dominant factors responsible for the hydrogenation and carbon chain propagation. The simple microkinetic model built on the basis of the above results shows that CH2 is the dominant CHx species on the surface in the temperature range of 300–800 K. Starting from a high coverage of CH2, the building block of the C-chain, CH2CH2 forms via a coupling reaction and then desorbs from the surface. CH2CH, appearing as the precursor, mainly forms from the coupling of CH2 + CH followed by CH2 insertion leading to CH2CHCH3. Although CH is more likely responsible for the chain propagation than CH2 in view of energy barriers, its contribution suffers from its low coverage at the considered conditions. These results are in good agreement with the experimental results.
Co-reporter:Yan-Ni Liang, Xia Yang, Songdong Ding, Shoujian Li, Fan Wang, Zhifang Chai and Dongqi Wang
New Journal of Chemistry 2015 vol. 39(Issue 10) pp:7716-7729
Publication Date(Web):02 Sep 2015
DOI:10.1039/C5NJ01285F
To assess the role of the lateral triazine group of 2,6-bis(1,2,4-triazin-3-yl) pyridine (BTP) when coordinated to Am(III), three tridentate N-donor ligands, i.e. BTP, 6-(-2-pyridyl)-2-pyridyl (hemi-BTP), and 2,2′:6′2′′-terpyridine (TPY), have been used to construct coordination complexes with Am(III), and the structures and binding modes of these complexes have been investigated using the B3LYP functional. The 1:1 and 1:2 (metal:ligand) type complexes, based on our calculations, form mainly via reactions Am(H2O)3(NO3)3 + L → AmL(NO3)3 + 3H2O and [Am(H2O)6(NO3)2]+ + 2L → [AmL2(NO3)2]+ + 6H2O. The Gibbs free energy changes were in the order of TPY > hemi-BTP > BTP, independent of the presence of nitrate ions in the complexes. We show that in 1:1 type complexes substitution of electron-donating groups to the three ligands can enhance their binding ability. From analysis of NPA charge and Mayer Bond Order, it is found that the value of binding free energy is correlated with charge transfers between the central metal and the ligand: the larger the ligand-to-metal charge transfer, the more negative the binding energy, and meanwhile, the smaller the Mayer bond order of the Am–N bonds. This suggests that the interaction between Am(III) and the tridentate ligands has a strong ionic feature, which is confirmed by the quantum theory of atoms-in-molecules (QTAIM) topological analysis. According to our calculations, the presence of the triazine group in BTP and hemi-BTP does not improve the binding affinity of the ligand to Am(III), compared to TPY, but facilitates the ligand to adopt a conformation that favors to coordinate with Am3+ than others via a dynamic isomerization process, and the electron-donating groups on the triazine group may enhance the charge transfer between Am(III) and the ligand, and thus stabilize the complex. We tentatively propose that the facile conversion between the conformations of BTP, which is more difficult for TPY and hemi-BTP, may significantly contribute to its higher affinity towards binding with Am(III).
Co-reporter:Xia Yang, Yanni Liang, Songdong Ding, Shoujian Li, Zhifang Chai, and Dongqi Wang
Inorganic Chemistry 2014 Volume 53(Issue 15) pp:7848-7860
Publication Date(Web):July 11, 2014
DOI:10.1021/ic500138w
The present theoretical study provides a realistic evaluation of the equilibrium structure, reaction modes, and bonding characteristics of a variety of neptunyl complexes formed with bis(triazinyl) N-donor extractants, which differ in their bridging groups such as pyridine, bipyridines, and orthophenanthroline, corresponding to the ligands (L) of tridentate bis(triazinyl)pyridines and tetradentate bis(triazinyl)bipyridines and bis(triazinyl)-1,10-phenanthrolines (BTPhens), respectively. Our calculations show that coordination of [NpO2]+ to tetradentate ligands is more favorable than that to tridentate ones no matter in a gas, aqueous, or organic phase. The presence of nitrate ions can enhance the coordination ability of neptunyl and stabilize the neutral NpO2L(NO3) complexes in thermodynamics. Our studies indicate that the complexation reaction mode [NpO2(H2O)n]+ + L + NO3– → NpO2L(NO3) + nH2O is the most probable at the interface between water and the organic phase. The contribution of an orthophenanthroline bridging group is relatively more pronounced compared to its pyridine counterpart in ligand-exchange reaction. Complexation reactions of hydrated neptunyl with C2-BTPhen and BTPhen assisted by a nitrate ion are favorable thermodynamically, resulting from the least deformation of the ligand and strong complexation stability. The quantum theory of atoms-in-molecules and charge decomposition analysis suggest that electron delocalization and charge transfer are the main reasons responsible for stabilization of the tetradentate complexes and reveal a strong ionic feature of the Np–ligand bonds. Inspection of the frontier molecular orbitals reveals a distinct 5f orbital (Np) interaction with ligand atoms, implying the extent of f-based covalency. Our study may facilitate the rational design of ligands toward the improvement of their binding ability with NpV and more efficient separation of Np in spent nuclear fuels.
Co-reporter:Jinghui Zeng, Xia Yang, Jiali Liao, Ning Liu, Yuanyou Yang, Zhifang Chai and Dongqi Wang
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 31) pp:16536-16546
Publication Date(Web):28 May 2014
DOI:10.1039/C4CP01381F
Density functional theory has been used to study the geometries and relative stabilities of the complexes of NpO2+ with the title compounds (L), including TMOGA, deprotonated N,N′-dimethyl-3-oxa-glutaramic acid (DMOGA) and their deprotonated oxydiacetic analog (ODA). Our calculations suggest that the complexes where the ligands appear as tridentate chelators are more stable than as bidentate ones, and the substitution of the amide group by carboxylate favors the formation of the complexes. Thermodynamically the 1:2 complex (Np–L2) is more favorable than the 1:1 complex (Np–L) in the cases of TMOGA and DMOGA, but not for the ODA anion. Taking into account the solvation effect of water, the 1:2 complex is more favorable than the 1:1 complex for all of the three ligands, though the reaction enthalpy decreases compared to that in the gas phase, and the formation of Np–(TMOGA)2 from Np–TMOGA is roughly a thermal neutral process. The strength of the NpO bond is weakened upon the coordination of ligands to Np(V) and the increase of the negative charge on the ligand (−1e for deprotonated DMOGA and −2e for deprotonated ODA). The Quantum Theory of Atoms-in-Molecules (QTAIM) was used here to analyze the bonding mode of NpO2+–Lx (x = 1, 2) and to compare the bond order data.
Co-reporter:Wanjian Ding and Dongqi Wang
Organometallics 2014 Volume 33(Issue 24) pp:7007-7010
Publication Date(Web):December 9, 2014
DOI:10.1021/om500797h
The mechanisms of CO2 insertion into the U–N bond of a silylamido NHC (N-heterocyclic carbene) U(III) complex were investigated theoretically. An earlier reported mechanism involving a reversible NHC carboxylation was found to be energetically too demanding, and a novel four-step mechanism featuring a direct CO2 insertion into the U–N bond is proposed.
Co-reporter:Peng Lian, Jue Li, Dong-Qi Wang, and Dong-Qing Wei
The Journal of Physical Chemistry B 2013 Volume 117(Issue 26) pp:7849-7856
Publication Date(Web):June 6, 2013
DOI:10.1021/jp312107r
The relevance of the pathway through which the second proton is delivered to the active site of P450cam and the subsequent coupling/uncoupling reactions has been investigated using Car–Parrinello molecular dynamics/molecular mechanics (CPMD/MM) dynamics simulations. Five models have been prepared, representing delivery pathways in the wild-type enzyme and its mutants in which Thr252 mutated into other residues with different side-chain length and hydrophobicity. In the simulations, coupling reaction is observed in the wild-type enzyme (Model A) and its T252S mutant (Model B), while the uncoupling products are obtained in the other three models (C, D, and E). Different from previous studies, a dynamic process of the last stage of coupling/uncoupling was observed. We found that the peroxide bond cleavage in coupling, the Fe–O bond stretching in uncoupling, proton transfer, and electron delivery take place spontaneously. Moreover, besides the intrinsic chemical differences between the two peroxide oxygen atoms, water molecules in the active site and the proton transfer pathway may play an important role in the determination of coupling/uncoupling. We conclude that by maintaining a specific proton transfer channel, Asp251–Thr252 channel, the wild-type enzyme could efficiently deliver the second proton to the ideal position for coupling reaction.
Co-reporter:Dongqi Wang, Anja Böckmann, Jožica Dolenc, Beat H. Meier, and Wilfred F. van Gunsteren
The Journal of Physical Chemistry B 2013 Volume 117(Issue 39) pp:11433-11447
Publication Date(Web):September 2, 2013
DOI:10.1021/jp400655v
NMR experiments have shown that water molecules in the crystal of the protein Crh are still mobile at temperatures well below 273 K. In order to investigate this water anomaly, a molecular dynamics (MD) simulation study of crystalline Crh was carried out to determine the mobility of water in this crystal. The simulations were carried out at three temperatures, 150, 200, and 291 K. Simulations of bulk water at these temperatures were also done to obtain the properties of the simple point charge (SPC) water model used at these temperatures and to allow a comparison of the properties of water in the Crh crystal with those of bulk water at the same temperatures. According to the simulations, water is immobilized at 150 K both in crystal and in bulk water. As expected, at 291 K it diffuses and rotates more slowly in the protein crystal than in bulk water. However, at 200 K, the translational and rotational mobility of the water molecules is larger in the crystal than in bulk water. The enhancement of water mobility in the crystal at 200 K was further investigated by MD simulations in which the backbone or all protein atoms were positionally restrained, and in which additionally the electrostatic protein-water interactions were removed. Of these changes in the environment of the water molecules, rigidifying the protein backbones slightly enhanced water diffusion, while it slowed down rotation. In contrast, removal of electrostatic protein–water interactions did not change water diffusion but enhanced rotational motion significantly. Further investigations are required to delineate particular features of the protein crystal that induce the anomalous behavior of water at 200 K.
Co-reporter:Dongqi Wang, Wilfred F. van Gunsteren and Zhifang Chai
Chemical Society Reviews 2012 vol. 41(Issue 17) pp:5836-5865
Publication Date(Web):09 Jul 2012
DOI:10.1039/C2CS15354H
We briefly review advances in computational actinoid (An) chemistry during the past ten years in regard to two issues: the geometrical and electronic structures, and reactions. The former addresses the An–O, An–C, and M–An (M is a metal atom including An) bonds in the actinoid molecular systems, including actinoid oxo and oxide species, actinoid–carbenoid, dinuclear and diatomic systems, and the latter the hydration and ligand exchange, the disproportionation, the oxidation, the reduction of uranyl, hydroamination, and the photolysis of uranium azide. Concerning their relevance to the electronic structures and reactions of actinoids and their importance in the development of an advanced nuclear fuel cycle, we also mentioned the work on actinoid carbides and nitrides, which have been proposed to be candidates of the next generation of nuclear fuel, and the oxidation of PuOx, which is important to understand the speciation of actinoids in the environment, followed by a brief discussion on the urgent need for a heavier involvement of computational actinoid chemistry in developing advanced reprocessing protocols of spent nuclear fuel. The paper is concluded with an outlook.
Co-reporter:Wanjian Ding, Weihai Fang, Zhifang Chai, and Dongqi Wang
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 10) pp:3605-3617
Publication Date(Web):August 7, 2012
DOI:10.1021/ct300075n
We report our recent DFT mechanistic study on the functionalization of CO2 and CS2 promoted by a trivalent uranium complex (Tp*)2UCH2Ph. In the calculations, the uranium atom is described by a quasi-relativistic 5f-in-core ECP basis set (LPP) developed for the trivalent uranium cation, which was qualified by the calculations with a quasi-relativistic small core ECP basis set (SPP) for uranium. According to our calculations, the functionalization proceeds in a stepwise manner, and the CO2 or CS2 does not interact with the central uranium atom to form a stable complex prior to the reaction due to the steric hindrance from the bulky ligands but directly cleaves the U–C (benzyl) bond by forming a C–C covalent bond. The released coordination site of uranium is concomitantly occupied by one chalcogen atom of the incoming molecule and gives an intermediate with the uranium atom interacting with the functionalized CO2 or CS2 in an η1 fasion. This step is followed by a reorientation of the (dithio)carboxylate side chain of the newly formed PhCH2CE2– (E = O, S) ligand to give the corresponding product. Energetically, the first step is characterized as the rate-determining step with a barrier of 9.5 (CO2) or 25.0 (CS2) kcal/mol, and during the reaction, the chalcogen atoms are reduced, while the methylene of the benzyl group is oxidized. Comparison of the results from SPP and LPP calculations indicates that our calculations qualify the use of an LPP treatment of the uranium atom toward a reasonable description of the model systems in the present study.
Co-reporter:Xin Liu, Lin Li, Bo Liu, Dongqi Wang, Yuliang Zhao, and Xingfa Gao
The Journal of Physical Chemistry A 2012 Volume 116(Issue 47) pp:11651-11655
Publication Date(Web):November 7, 2012
DOI:10.1021/jp306481e
Despite its experimental characterization, the detailed geometry and electronic structure of actinide metallofullerene U@C82 have been rarely studied. We predict that #5C82 and #8C82 are the best cages for the encapsulation of monovalent and tetravalent U (i.e., U+ and U4+), respectively; while #9C82 is the best cage for divalent, trivalent, pentavalent, and hexavalent U cations (i.e., U2+, U3+, U5+, and U6+). U@#9C82 is the thermodynamically most stable one among all the isomers and thus corresponds to the most experimentally isolable isomer of U@C82. The calculated spin density explicitly suggests that the endohedral metallofullerene U@#9C82 is a trivalent ion-pair with an electronic configuration of U3+@C823–. The proposed geometry and electronic structure of U3+@#9C823– are in good agreement with the experimental observation.
Co-reporter:Dongqi Wang, Wilfred F. van Gunsteren and Zhifang Chai
Chemical Society Reviews 2012 - vol. 41(Issue 17) pp:NaN5865-5865
Publication Date(Web):2012/07/09
DOI:10.1039/C2CS15354H
We briefly review advances in computational actinoid (An) chemistry during the past ten years in regard to two issues: the geometrical and electronic structures, and reactions. The former addresses the An–O, An–C, and M–An (M is a metal atom including An) bonds in the actinoid molecular systems, including actinoid oxo and oxide species, actinoid–carbenoid, dinuclear and diatomic systems, and the latter the hydration and ligand exchange, the disproportionation, the oxidation, the reduction of uranyl, hydroamination, and the photolysis of uranium azide. Concerning their relevance to the electronic structures and reactions of actinoids and their importance in the development of an advanced nuclear fuel cycle, we also mentioned the work on actinoid carbides and nitrides, which have been proposed to be candidates of the next generation of nuclear fuel, and the oxidation of PuOx, which is important to understand the speciation of actinoids in the environment, followed by a brief discussion on the urgent need for a heavier involvement of computational actinoid chemistry in developing advanced reprocessing protocols of spent nuclear fuel. The paper is concluded with an outlook.
Co-reporter:Xia Yang, Zhifang Chai and Dongqi Wang
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 11) pp:NaN7547-7547
Publication Date(Web):2015/02/12
DOI:10.1039/C4CP04586F
Four types of reaction mechanisms for the oxo ligand exchange of monomeric and dimeric neptunyl(VI) hydroxide in aqueous solution were explored computationally using density functional theory (DFT) and ab initio classical molecular dynamics. The obtained results were compared with previous studies on the oxo exchange of uranyl hydroxide, as well as with experiments. It is found that the stable T-shaped [NpO3(OH)3]3− intermediate is a key species for oxo exchange in the proton transfer in mononuclear Path I and binuclear Path IV, similar to the case of uranyl(VI) hydroxide. Path I is thought to be the preferred oxo exchange mechanism for neptunyl(VI) hydroxide in our calculations, due to the lower activation energy (22.7 and 13.1 kcal mol−1 for ΔG‡ and ΔH‡, respectively) of the overall reaction. Path II via a cis-neptunyl structure assisted by a water molecule might be a competitive channel against Path I with a mononuclear mechanism, owing to a rapid dynamical process occurring in Path II. In Path IV with the binuclear mechanism, oxo exchange is accomplished via the interaction between [NpO2(OH)4]2− and T-shaped [NpO3(OH)3]3− with a low activation energy for the rate-determining step, however, the overall energy required to fulfill the reaction is slightly higher than that in mononuclear Path I, suggesting a possible binuclear process in the higher energy region. The chemical bonding evolution along the reaction pathways was discussed by using topological methodologies of the electron localization function (ELF).
Co-reporter:Hui Wang, Zhifang Chai and Dongqi Wang
Dalton Transactions 2015 - vol. 44(Issue 4) pp:NaN1654-1654
Publication Date(Web):2014/11/12
DOI:10.1039/C4DT02872D
The adsorption of [UO2(H2O)5]2+ on a hydroxylated α-SiO2(001) surface was studied by periodic density functional theory (DFT) and ab initio molecular dynamics (AIMD) simulation. The effects of pH, CO2, aqua solution and anionic ligands (OH−, NO3− and Cl−) on the adsorption geometry and stability were investigated. The results show that the adsorption of uranyl on a hydroxylated α-SiO2(001) surface leads to the formation of inner-sphere complexes, in which the bidentate complex at the double deprotonated site is most favored. The binding strengths of bidentate and monodentate complexes at the same site are similar, and they become weaker as the number of protons increases at the adsorption site, indicating an enhancement of the adsorption strength at higher pH values within a certain range. Strong chemical interaction plays an important role in all inner-sphere complexes. The hydrogen bonds are formed between uranyl and the hydroxylated surface in all inner- and outer-sphere complexes. The presence of CO2 weakens the adsorption of uranyl on the surface by forming uranyl carbonate (CO32−, HCO3−) complexes. The effect of the anion ligands depends on their charged state and their concentration in solutions. The explicit treatment of water environment in the models has a slight effect on the adsorption configuration. These results are consistent with the experimental observations.
Co-reporter:Jinghui Zeng, Xia Yang, Jiali Liao, Ning Liu, Yuanyou Yang, Zhifang Chai and Dongqi Wang
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 31) pp:NaN16546-16546
Publication Date(Web):2014/05/28
DOI:10.1039/C4CP01381F
Density functional theory has been used to study the geometries and relative stabilities of the complexes of NpO2+ with the title compounds (L), including TMOGA, deprotonated N,N′-dimethyl-3-oxa-glutaramic acid (DMOGA) and their deprotonated oxydiacetic analog (ODA). Our calculations suggest that the complexes where the ligands appear as tridentate chelators are more stable than as bidentate ones, and the substitution of the amide group by carboxylate favors the formation of the complexes. Thermodynamically the 1:2 complex (Np–L2) is more favorable than the 1:1 complex (Np–L) in the cases of TMOGA and DMOGA, but not for the ODA anion. Taking into account the solvation effect of water, the 1:2 complex is more favorable than the 1:1 complex for all of the three ligands, though the reaction enthalpy decreases compared to that in the gas phase, and the formation of Np–(TMOGA)2 from Np–TMOGA is roughly a thermal neutral process. The strength of the NpO bond is weakened upon the coordination of ligands to Np(V) and the increase of the negative charge on the ligand (−1e for deprotonated DMOGA and −2e for deprotonated ODA). The Quantum Theory of Atoms-in-Molecules (QTAIM) was used here to analyze the bonding mode of NpO2+–Lx (x = 1, 2) and to compare the bond order data.

![1,2,4-Triazine, 3,3'-[2,2'-bipyridine]-6,6'-diylbis-](http://img.cochemist.com/ccimg/887000/886971-07-9.png)
![1,2,4-Triazine, 3,3'-[2,2'-bipyridine]-6,6'-diylbis-](http://img.cochemist.com/ccimg/887000/886971-07-9_b.png)
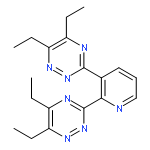
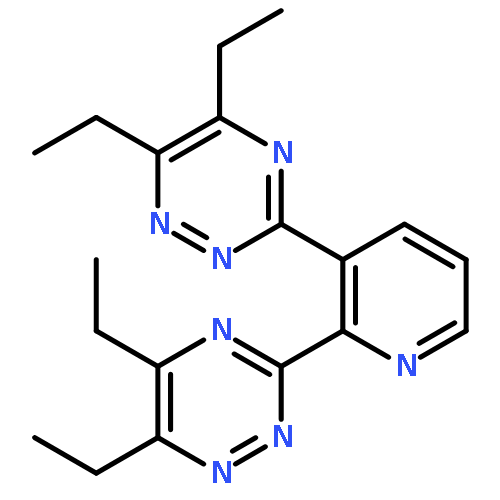
![3-[6-(1,2,4-TRIAZIN-3-YL)PYRIDIN-2-YL]-1,2,4-TRIAZINE](http://img.cochemist.com/ccimg/220600/220525-55-3.png)
![3-[6-(1,2,4-TRIAZIN-3-YL)PYRIDIN-2-YL]-1,2,4-TRIAZINE](http://img.cochemist.com/ccimg/220600/220525-55-3_b.png)
![3-[6-(5,6-DIETHYL-1,2,4-TRIAZIN-3-YL)PYRIDIN-2-YL]-5,6-DIETHYL-1,2,4-TRIAZINE](http://img.cochemist.com/ccimg/216000/215917-62-7.png)
![3-[6-(5,6-DIETHYL-1,2,4-TRIAZIN-3-YL)PYRIDIN-2-YL]-5,6-DIETHYL-1,2,4-TRIAZINE](http://img.cochemist.com/ccimg/216000/215917-62-7_b.png)















