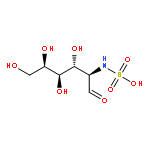
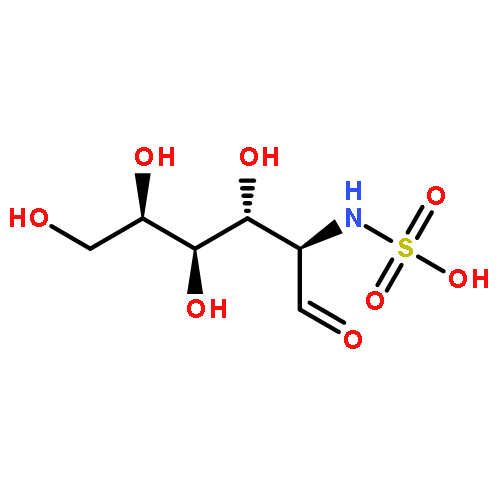
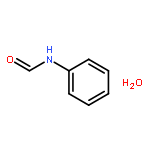
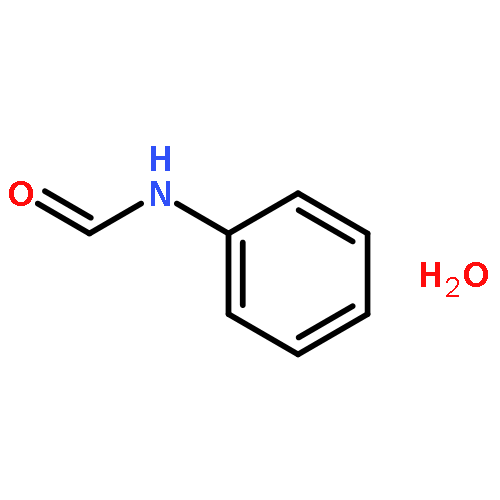
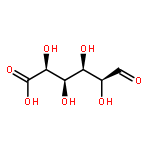
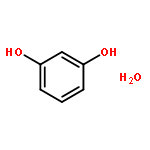
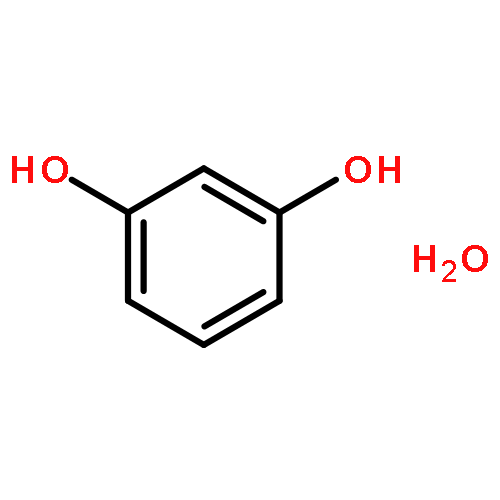
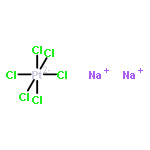
Co-reporter:Rosaria Cercola, Edward Matthews, and Caroline E. H. Dessent
The Journal of Physical Chemistry B June 8, 2017 Volume 121(Issue 22) pp:5553-5553
Publication Date(Web):May 18, 2017
DOI:10.1021/acs.jpcb.7b03435
We report the first UV laser photodissociation spectra (4.0–5.8 eV) of gas-phase deprotonated adenosine 5′-triphosphate, diphosphate and monophosphate anions. The photodepletion spectra of these anions display strong absorption bands across the region of 4.6–5.2 eV, consistent with excitation of a primarily adenine-centered π–π* transition. The spectra appear insensitive to the charge of the species (i.e., the spectrum of [ATP-2H]2– closely resembles that of [ATP-H]−), while the spectral profile is affected to a greater extent by the variation of the molecular structure, i.e. the [AMP-H]− and [ADP-H]− photodepletion spectra display similar profiles while the [ATP-H]− spectrum is distinctive. The photodepletion cross-section also decreases for the ATP anions compared to both the AMP and ADP anions, reflecting a high intrinsic photostability of ATP versus both AMP and ADP. A range of photofragments are produced across the 4.0–5.8 eV spectral range for all of the ATP analogues studied. These fragments are primarily associated with fragmentation on the ground-state electronic surface, indicative of a statistical decay process where ultrafast decay is followed by ergodic dissociation. However, while the photofragments observed following photoexcitation of the monoanionic species, [AMP-H]− to [ADP-H]− to [ATP-H]− are entirely consistent with statistical decay, an additional group of photofragments are observed for the dianionic species, [ADP-2H]2– and [ATP-2H]2–, that we associate with electron detachment, and subsequent fragmentation of the resulting electron-detached photofragment. TDDFT calculations are presented to support the interpretation of the experimental data, and confirm that the electronic structure of the adenine moiety is relatively unperturbed by varying the overall charge.
Co-reporter:Edward Matthews
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 26) pp:17434-17440
Publication Date(Web):2017/07/05
DOI:10.1039/C7CP02817B
The identification of protonation sites in electrosprayed molecules remains a challenge in contemporary physical science. We present the first demonstration that low-resolution, UV laser photodissociation spectroscopy can be applied in situ to identify the protomers of para-aminobenzoic acid (PABA) formed via electrospray from a single solution. Electronic absorption spectra are recorded via photodepletion and photofragmentation for PABA electrosprayed from solutions of water and acetonitrile. Using this approach, two protomers can be straightforwardly identified, with only the carboxylic acid protomer being produced on electrospray from water while the amine-protonated isomer dominates upon electrospray from acetonitrile. High-level SORCI and MRCI calculations are presented to provide insight into the origin of the distinctive electronic spectra displayed by the protomers. Our results are in excellent agreement with previous PABA studies conducted using established techniques, and demonstrate that UV photodissociation spectroscopy of electrosprayed ions has potential as a new diagnostic tool for identifying protomeric species.
Co-reporter:Edward Matthews, Ananya Sen, Naruo Yoshikawa, Ed Bergström and Caroline E. H. Dessent
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 22) pp:15143-15152
Publication Date(Web):12 May 2016
DOI:10.1039/C6CP01676F
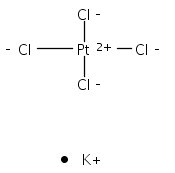
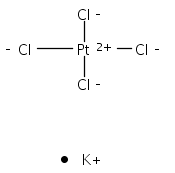
Isolated molecular clusters of adenine, cytosine, thymine and uracil bound to hexachloroplatinate, PtCl62−, have been studied using laser electronic photodissociation spectroscopy to investigate photoactivation of a platinum complex in the vicinity of a nucleobase. These metal complex–nucleobase clusters represent model systems for identifying the fundamental photochemical processes occurring in photodynamic platinum drug therapies that target DNA. This is the first study to explore the specific role of a strongly photoactive platinum compound in the aggregate complex. Each of the clusters studied displays a broadly similar absorption spectra, with a strong λmax ∼ 4.6 eV absorption band and a subsequent increase in the absorption intensity towards higher spectral-energy. The absorption bands are traced to ligand-to-metal-charge-transfer excitations on the PtCl62− moiety within the cluster, and result in Cl−·nucleobase and PtCl5− as primary photofragments. These results demonstrate how selective photoexcitation can drive distinctive photodecay channels for a model photo-pharmaceutical. In addition, cluster absorption due to excitation of nucleobase-centred chromophores is observed in the region around 5 eV. For the uracil cluster, photofragments consistent with ultrafast decay of the excited state and vibrational predissociation on the ground-state surface are observed. However, this decay channel becomes successively weaker on going from thymine to cytosine to adenine, due to differential coupling of the excited states to the electron detachment continuum. These effects demonstrate the distinctive photophysical characteristics of the different nucleobases, and are discussed in the context of the recently recorded photoelectron spectra of theses clusters.
Co-reporter:Edward Matthews and Caroline E. H. Dessent
The Journal of Physical Chemistry A 2016 Volume 120(Issue 46) pp:9209-9216
Publication Date(Web):November 1, 2016
DOI:10.1021/acs.jpca.6b10433
Even in relatively simple molecules, the sites of protonation or deprotonation formed upon electrospray ionization can be controversial. This situation means that it is important to develop new approaches for identifying “protomers” and “deprotomers”. In this study, we demonstrate that routine, low-resolution UV laser photodissociation spectroscopy can be applied to identify the gaseous protomers of nicotinamide formed upon electrospray. Nicotinamide is an important biological molecule that possesses multiple protonation sites associated with its pyridine and amide groups. We obtain a gas-phase absorption spectrum for protonated nicotinamide that closely resembles the solution-phase spectrum. However, photoexcitation of protonated nicotinamide produces numerous ionic photofragments, and the spectral profiles for production of these photofragments from protonated nicotinamide reveal the existence of two distinctive chromophores, which can be traced to the existence of pyridine and amide protomers. We observe that these protomers are associated with absorption bands centered at 4.96 and 4.73 eV, respectively, with the protomers appearing in an approximate ratio of ∼2:1. The fact that the considerably less favorable amide protomer is observed in substantial quantities in the gas phase is surprising given that the pyridine protomer is the lower-energy species in both solution and gas phase. The high amounts of amide protomers observed here can be explained as arising from asymmetric pyridine protomer–amide bound dimers, present in solution or in the electrosprayed droplets, which lead to enhanced formation of the unexpected amide-protonated isomers.
Co-reporter:Ananya Sen, Gao-Lei Hou, Xue-Bin Wang, and Caroline E. H. Dessent
The Journal of Physical Chemistry B 2015 Volume 119(Issue 35) pp:11626-11631
Publication Date(Web):August 5, 2015
DOI:10.1021/acs.jpcb.5b07108
We report the first low-temperature photoelectron spectra of isolated gas-phase complexes of the platinum II cyanide dianion bound to nucleobases. These systems are models for understanding platinum-complex photodynamic therapies, and a knowledge of the intrinsic photodetachment properties is crucial for characterizing their broader photophysical properties. Well-resolved, distinct peaks are observed in the spectra, consistent with complexes where the Pt(CN)42– moiety is largely intact. Adiabatic electron detachment energies for the dianion-nucleobase complexes are measured to be 2.39–2.46 eV. The magnitudes of the repulsive Coulomb barriers of the complexes are estimated to be between 1.9 and 2.1 eV, values that are lower than for the bare Pt(CN)42– dianion as a result of charge solvation by the nucleobases. In addition to the resolved spectral features, broad featureless bands indicative of delayed electron detachment are observed in the 193 nm photoelectron spectra of the four dianion-nucleobase complexes and also in the 266 nm spectra of the Pt(CN)42–·thymine and Pt(CN)42–·adenine complexes. The selective excitation of these features in the 266 nm spectra is attributed to one-photon excitation of [Pt(CN)42–·thymine]* and [Pt(CN)42–·adenine]* long-lived excited states that can effectively couple to the electron detachment continuum, producing strong electron detachment signals. We attribute the delayed electron detachment bands observed here for Pt(CN)42–·thymine and Pt(CN)42–·adenine but not for Pt(CN)42–·uracil and Pt(CN)42–·cytosine to fundamental differences in the individual nucleobase photophysics following 266 nm excitation. This indicates that the Pt(CN)42– dianion in the clusters can be viewed as a “dynamic tag” which has the propensity to emit electrons when the attached nucleobase displays a long-lived excited state.
Co-reporter:Ananya Sen, Thomas F. M. Luxford, Naruo Yoshikawa and Caroline E. H. Dessent
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 29) pp:15490-15500
Publication Date(Web):16 Jun 2014
DOI:10.1039/C4CP00989D
Isolated molecular clusters of adenine, cytosine, thymine and uracil with Pt(CN)62− and Pt(CN)42− were studied for the first time to characterize the binding and reactivity of isolated transition metal complex ions with nucleobases. These clusters represent model systems for understanding metal complex–DNA adducts, as a function of individual nucleobases. Collisional excitation revealed that the clusters decay on the ground electronic surface by either solvent evaporation (i.e. loss of a nucleobase unit from the cluster) or via proton transfer from the nucleobase to the dianion. The Pt(CN)62−–nucleobase clusters decay only by solvent evaporation, while the Pt(CN)42− clusters fragment by both pathways. The enhanced proton-transfer reactivity of Pt(CN)42− is attributed to the higher charge-density of the ligands in this transition metal anion. % fragmentation curves of the clusters reveal that the adenine clusters display distinctively higher fragmentation onsets, which are traced to the propensity of adenine to form the shortest intercluster H-bond. We also present laser electronic photodissociation measurements for the Pt(CN)62−·Ur, Pt(CN)42−·Ur and Pt(CN)42−·Ur2 clusters to illustrate the potential of exploring metal complex DNA photophysics as a function of nucleobase within well-defined gaseous clusters. The spectra reported herein represent the first such measurements. We find that the electronic excited states decay with production of the same fragments (associated with solvent evaporation and proton transfer) observed upon collisional excitation of the electronic ground state, indicating ultrafast deactivation of the excited-state uracil-localized chromophore followed by vibrational predissociation.
Co-reporter:Ananya Sen and Caroline E. H. Dessent
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 19) pp:3281-3285
Publication Date(Web):September 9, 2014
DOI:10.1021/jz501749j
We report the first UV laser spectroscopic study of isolated gas-phase complexes of platinum metal complex anions bound to a nucleobase as model systems for exploring at the molecular level the key photophysical processes involved in photodynamic therapy. Spectra of the PtIV(CN)62–·Ur and PtII(CN)42–·Ur complexes were acquired across the 220–320 nm range using mass-selective photodepletion and photofragment action spectroscopy. The spectra of both complexes reveal prominent UV absorption bands (λmax = 4.90 and 4.70 eV) that we assign primarily to excitation of the Ur π–π* localized chromophore. Distinctive UV photofragmentation products are observed for the complexes, with PtIV(CN)62–·Ur photoexcitation resulting in complex fission, while PtII(CN)42–·Ur photoexcitation initiates a nucleobase proton-transfer reaction across 4.4–5.2 eV and electron detachment above 5.2 eV. The observed photofragments are consistent with ultrafast decay of a Ur localized excited state back to the electronic ground state followed by intramolecular vibrational relaxation and ergodic complex fragmentation.Keywords: laser photodissociation; multiply charged anions; nucleobase photochemistry; photodynamic therapy; Pt complex photochemistry; uracil photophysics;
Co-reporter:Thomas F.M. Luxford, Edward M. Milner, Naruo Yoshikawa, Chad Bullivant, Caroline E.H. Dessent
Chemical Physics Letters 2013 Volume 577() pp:1-5
Publication Date(Web):9 July 2013
DOI:10.1016/j.cplett.2013.05.040
•First study of isolated carboxylate–amino acid interactions as function of anion size.•Dicarboxylic acid–Arg clusters display structural change with increasing chain length.•Carboxylate–Arg clusters display evidence of anion-stabilized zwitterionic amino acid.Complexation of deprotonated carboxylic acids with arginine was investigated using collision-induced dissociation to probe the nature of isolated carboxylate–amino acid interactions as a function of anion size. Monocarboxylic CH3(CH2)nCOO−·Arg (n = 3–7, 9, 10) and dicarboxylic acid COOH(CH2)nCOO−·Arg (n = 3–5, 7–10) complexes were investigated. For the dicarboxylic acid clusters, chain length has a significant effect on the %fragmentation energies with the n = 9, 10 systems fragmenting at significantly lower energies than the corresponding shorter chain systems. Molecular mechanics calculations suggest that this fragmentation energy shift is associated with the longer-chain dicarboxylic acid–Arg clusters switching to ring structures.Graphical abstract
Co-reporter:Martin Walker, Ananya Sen, Andrew J.A. Harvey, Caroline E.H. Dessent
Chemical Physics Letters 2013 Volume 588() pp:43-46
Publication Date(Web):19 November 2013
DOI:10.1016/j.cplett.2013.09.074
Highlights
- •
Br− can stabilize the zwitterionic form of arginine in an isolated cluster.
- •
Zwitterionic and canonical minima are very close in energy for Br−·Arg.
- •
The canonical tautomer is lower in energy for the Cl−·Arg cluster.
- •
Conformation is crucial in determining canonical vs zwitterion relative energies.
- •
Comparison to experiment suggests that messenger tagged IR experiments are vital.
Co-reporter:Martin Walker, Andrew J. A. Harvey, Ananya Sen, and Caroline E. H. Dessent
The Journal of Physical Chemistry A 2013 Volume 117(Issue 47) pp:12590-12600
Publication Date(Web):October 22, 2013
DOI:10.1021/jp408166m
We present a comparative assessment of the performance of the M06 suite of density functionals (M06, M06-2X, and M06-HF) against an MP2 benchmark for calculating the relative energies and geometric structures of the Cl–·arginine and Br–·arginine halide ion–amino acid clusters. Additional results are presented for the popular B3LYP density functional. The Cl–·arginine and Br–·arginine complexes are important prototypes for the phenomenon of anion-induced zwitterion formation. Results are presented for the canonical (noncharge separated) and zwitterionic (charge separated) tautomers of the clusters, as well as the numerous conformational isomers of the clusters. We find that all of the M06 functions perform well in terms of predicting the general trends in the conformer relative energies and identifying the global minimum conformer. This is in contrast to the B3LYP functional, which performed significantly less well for the canonical tautomers of the clusters where dispersion interactions contribute more significantly to the conformer energetics. We find that the M06 functional gave the lowest mean unsigned error for the relative energies of the canonical conformers (2.10 and 2.36 kJ/mol for Br–·arginine and Cl–·arginine), while M06-2X gave the lowest mean unsigned error for the zwitterionic conformers (0.85 and 1.23 kJ/mol for Br–·arginine and Cl–·arginine), thus providing insight into the types of physical systems where each of these functionals should perform best.
Co-reporter:Edward M. Milner, Michael G. D. Nix, and Caroline E. H. Dessent
The Journal of Physical Chemistry A 2012 Volume 116(Issue 2) pp:801-809
Publication Date(Web):December 21, 2011
DOI:10.1021/jp208183p
We report the first low-energy collisional-induced dissociation studies of the X–·arginine (X– = F–, Cl–, Br–, I–, NO3–, ClO3–) series of clusters to investigate the novel phenomenom of anion-induced zwitterion formation in a gas-phase amino acid. Fragmentation of the small halide ion clusters (F–·arginine and Cl–·arginine) is dominated by deprotonation of the arginine, whereas the major fragmentation channel for the largest ion clusters (I–·arginine and ClO3–·arginine) corresponds to simple cluster fission into the ion and neutral molecule. However, the fragmentation profiles of Br–·arginine and NO3–·arginine, display distinctive features that are consistent with the presence of the zwitterionic form of the amino acid in these clusters. The various dissociation pathways have been studied as a function of % collision energy and are discussed in comparison to the fragmentation profiles of protonated and deprotonated arginine. Electronic structure calculations are presented for Br–·arginine to support the presence of the zwitterionic amino acid in this complex. The results obtained in this work provide important information on the low-energy potential energy surfaces of these anion–amino acid clusters and reveal the presence of several overlapping surfaces in the low-energy region for the Br–·arginine and NO3–·arginine systems.
Co-reporter:Ananya Sen, Thomas F. M. Luxford, Naruo Yoshikawa and Caroline E. H. Dessent
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 29) pp:NaN15500-15500
Publication Date(Web):2014/06/16
DOI:10.1039/C4CP00989D
Isolated molecular clusters of adenine, cytosine, thymine and uracil with Pt(CN)62− and Pt(CN)42− were studied for the first time to characterize the binding and reactivity of isolated transition metal complex ions with nucleobases. These clusters represent model systems for understanding metal complex–DNA adducts, as a function of individual nucleobases. Collisional excitation revealed that the clusters decay on the ground electronic surface by either solvent evaporation (i.e. loss of a nucleobase unit from the cluster) or via proton transfer from the nucleobase to the dianion. The Pt(CN)62−–nucleobase clusters decay only by solvent evaporation, while the Pt(CN)42− clusters fragment by both pathways. The enhanced proton-transfer reactivity of Pt(CN)42− is attributed to the higher charge-density of the ligands in this transition metal anion. % fragmentation curves of the clusters reveal that the adenine clusters display distinctively higher fragmentation onsets, which are traced to the propensity of adenine to form the shortest intercluster H-bond. We also present laser electronic photodissociation measurements for the Pt(CN)62−·Ur, Pt(CN)42−·Ur and Pt(CN)42−·Ur2 clusters to illustrate the potential of exploring metal complex DNA photophysics as a function of nucleobase within well-defined gaseous clusters. The spectra reported herein represent the first such measurements. We find that the electronic excited states decay with production of the same fragments (associated with solvent evaporation and proton transfer) observed upon collisional excitation of the electronic ground state, indicating ultrafast deactivation of the excited-state uracil-localized chromophore followed by vibrational predissociation.
Co-reporter:Edward Matthews and Caroline E. H. Dessent
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 26) pp:NaN17440-17440
Publication Date(Web):2017/06/19
DOI:10.1039/C7CP02817B
The identification of protonation sites in electrosprayed molecules remains a challenge in contemporary physical science. We present the first demonstration that low-resolution, UV laser photodissociation spectroscopy can be applied in situ to identify the protomers of para-aminobenzoic acid (PABA) formed via electrospray from a single solution. Electronic absorption spectra are recorded via photodepletion and photofragmentation for PABA electrosprayed from solutions of water and acetonitrile. Using this approach, two protomers can be straightforwardly identified, with only the carboxylic acid protomer being produced on electrospray from water while the amine-protonated isomer dominates upon electrospray from acetonitrile. High-level SORCI and MRCI calculations are presented to provide insight into the origin of the distinctive electronic spectra displayed by the protomers. Our results are in excellent agreement with previous PABA studies conducted using established techniques, and demonstrate that UV photodissociation spectroscopy of electrosprayed ions has potential as a new diagnostic tool for identifying protomeric species.
Co-reporter:Edward Matthews, Ananya Sen, Naruo Yoshikawa, Ed Bergström and Caroline E. H. Dessent
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 22) pp:NaN15152-15152
Publication Date(Web):2016/05/12
DOI:10.1039/C6CP01676F
Isolated molecular clusters of adenine, cytosine, thymine and uracil bound to hexachloroplatinate, PtCl62−, have been studied using laser electronic photodissociation spectroscopy to investigate photoactivation of a platinum complex in the vicinity of a nucleobase. These metal complex–nucleobase clusters represent model systems for identifying the fundamental photochemical processes occurring in photodynamic platinum drug therapies that target DNA. This is the first study to explore the specific role of a strongly photoactive platinum compound in the aggregate complex. Each of the clusters studied displays a broadly similar absorption spectra, with a strong λmax ∼ 4.6 eV absorption band and a subsequent increase in the absorption intensity towards higher spectral-energy. The absorption bands are traced to ligand-to-metal-charge-transfer excitations on the PtCl62− moiety within the cluster, and result in Cl−·nucleobase and PtCl5− as primary photofragments. These results demonstrate how selective photoexcitation can drive distinctive photodecay channels for a model photo-pharmaceutical. In addition, cluster absorption due to excitation of nucleobase-centred chromophores is observed in the region around 5 eV. For the uracil cluster, photofragments consistent with ultrafast decay of the excited state and vibrational predissociation on the ground-state surface are observed. However, this decay channel becomes successively weaker on going from thymine to cytosine to adenine, due to differential coupling of the excited states to the electron detachment continuum. These effects demonstrate the distinctive photophysical characteristics of the different nucleobases, and are discussed in the context of the recently recorded photoelectron spectra of theses clusters.