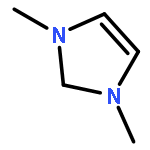
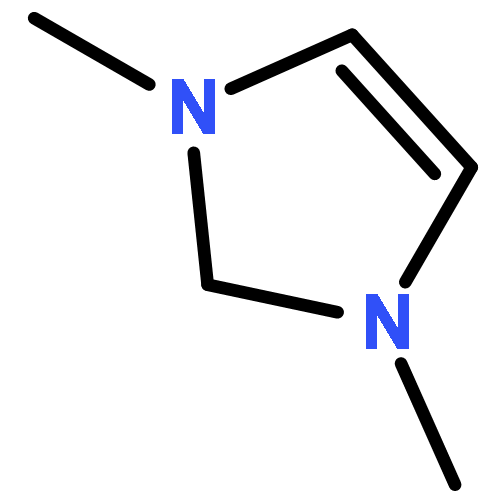
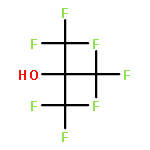
Co-reporter:Aaron J. Teator;Yuan Tian;Mu Chen; Jeehiun K. Lee; Christopher W. Bielawski
Angewandte Chemie 2015 Volume 127( Issue 39) pp:11721-11725
Publication Date(Web):
DOI:10.1002/ange.201506269
Abstract
The first isolable, photoswitchable N-heterocyclic carbene was synthesized and found to undergo reversible electrocyclic isomerization upon successive exposure to UV and visible radiation. The UV-induced ring closure afforded substantial changes to the electronic structure of the dithienylethene-based NHC, as evidenced by changes in the corresponding UV/Vis absorption and 13C NMR spectra. Likewise, molecular orbital calculations revealed diminished electron density at the carbene nucleus upon photocyclization, consistent with the enhanced electrophilicity displayed by the ring-closed form. The photoswitchable NHC was successfully switched between its ring-opened and ring-closed states with high fidelity over multiple cycles. Furthermore, the ring-closed isomer was found to undergo facile NH bond activation, allowing for the controlled capture and release of ammonia upon cycling between its isomeric states.
Co-reporter:Aaron J. Teator;Yuan Tian;Mu Chen; Jeehiun K. Lee; Christopher W. Bielawski
Angewandte Chemie International Edition 2015 Volume 54( Issue 39) pp:11559-11563
Publication Date(Web):
DOI:10.1002/anie.201506269
Abstract
The first isolable, photoswitchable N-heterocyclic carbene was synthesized and found to undergo reversible electrocyclic isomerization upon successive exposure to UV and visible radiation. The UV-induced ring closure afforded substantial changes to the electronic structure of the dithienylethene-based NHC, as evidenced by changes in the corresponding UV/Vis absorption and 13C NMR spectra. Likewise, molecular orbital calculations revealed diminished electron density at the carbene nucleus upon photocyclization, consistent with the enhanced electrophilicity displayed by the ring-closed form. The photoswitchable NHC was successfully switched between its ring-opened and ring-closed states with high fidelity over multiple cycles. Furthermore, the ring-closed isomer was found to undergo facile NH bond activation, allowing for the controlled capture and release of ammonia upon cycling between its isomeric states.
Co-reporter:Atanu Maiti ; Anna Zhachkina Michelson ; Cherece J. Armwood ; Jeehiun K. Lee ;Alexander C. Drohat
Journal of the American Chemical Society 2013 Volume 135(Issue 42) pp:15813-15822
Publication Date(Web):September 24, 2013
DOI:10.1021/ja406444x
5-Methylcytosine (mC) is an epigenetic mark that impacts transcription, development, and genome stability, and aberrant DNA methylation contributes to aging and cancer. Active DNA demethylation involves stepwise oxidation of mC to 5-hydroxymethylcytosine, 5-formylcytosine (fC), and potentially 5-carboxylcytosine (caC), excision of fC or caC by thymine DNA glycosylase (TDG), and restoration of cytosine via follow-on base excision repair. Here, we investigate the mechanism for TDG excision of fC and caC. We find that 5-carboxyl-2′-deoxycytidine ionizes with pKa values of 4.28 (N3) and 2.45 (carboxyl), confirming that caC exists as a monoanion at physiological pH. Calculations do not support the proposal that G·fC and G·caC base pairs adopt a wobble structure that is recognized by TDG. Previous studies show that N-glycosidic bond hydrolysis follows a stepwise (SN1) mechanism, and that TDG activity increases with pyrimidine N1 acidity, that is, leaving group quality of the target base. Calculations here show that fC and the neutral tautomers of caC are acidic relative to other TDG substrates, but the caC monoanion exhibits poor acidity and likely resists TDG excision. While fC activity is independent of pH, caC excision is acid-catalyzed, and the pH profile indicates that caC ionizes in the enzyme–substrate complex with an apparent pKa of 5.8, likely at N3. Mutational analysis reveals that Asn191 is essential for excision of caC but dispensable for fC activity, indicating that N191 may stabilize N3-protonated forms of caC to facilitate acid catalysis and suggesting that N191A-TDG could potentially be useful for studying DNA demethylation in cells.
Co-reporter:Kai Wang, Mu Chen, Qiaoyi Wang, Xiaodong Shi, and Jeehiun K. Lee
The Journal of Organic Chemistry 2013 Volume 78(Issue 14) pp:7249-7258
Publication Date(Web):June 28, 2013
DOI:10.1021/jo4012738
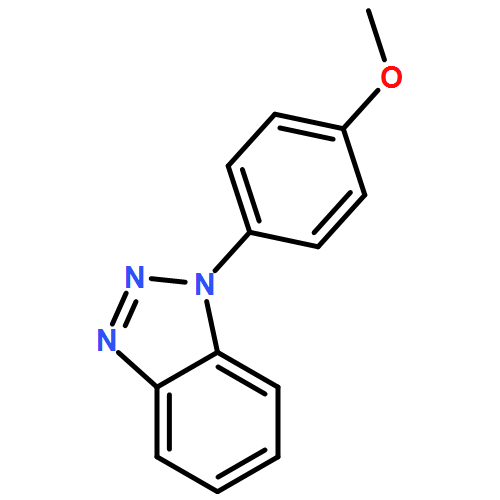
1,2,3-Triazoles have come to the forefront as compounds of import in a vast number of applications. The fundamental properties of these species, however, remain largely unknown. Herein, the gas phase properties of 4-phenyl-1,2,3-triazole, benzotriazole, and a series of 1-phenylbenzotriazoles are described. Proton affinity and acidity values are computed and measured. Furthermore, ion–molecule reactions and H/D exchange studies are used to ascertain tautomer prevalence for the 4-phenyl species.
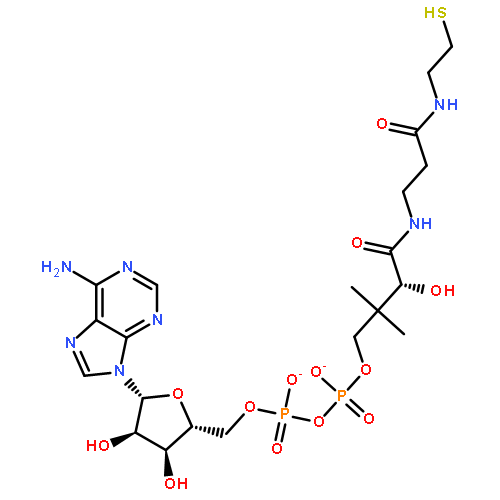
Co-reporter:Anna Zhachkina, Min Liu, Xuejun Sun, F. Sedinam Amegayibor and Jeehiun K. Lee
The Journal of Organic Chemistry 2009 Volume 74(Issue 19) pp:7429-7440
Publication Date(Web):September 4, 2009
DOI:10.1021/jo901479m
The gas phase acidity (ΔHacid and ΔGacid) and proton affinity (PA, and gas phase basicity (GB)) of adenine, guanine, and O6-methylguanine (OMG) have been examined using both theoretical (B3LYP/6-31+G*) and experimental (bracketing, Cooks kinetic) methods. We previously measured the acidity of adenine using bracketing methods; herein we measure the acidity of adenine by the Cooks kinetic method (ΔHacid = 335 ± 3 kcal mol−1; ΔGacid = 329 ± 3 kcal mol−1). We also measured the PA/GB of adenine using both bracketing and Cooks methods (PA = 224 and 225 kcal mol−1; GB = 216 and 217 kcal mol−1). Guanine is calculated to have several stable tautomers in the gas phase, in contrast to in solution, where the canonical tautomer predominates. Experimental measurements of gas phase guanine properties are difficult due to its nonvolatility; using electrospray and the Cooks kinetic method, we are able to measure a ΔHacid of 335 ± 3 kcal mol−1 (ΔGacid = 328 ± 3 kcal mol−1). The proton affinity is 227 ± 3 kcal mol−1 (GB = 219 ± 3 kcal mol−1). Comparison of these values to calculations indicates that we may have a mixture of the keto and enol tautomers under our conditions in the gas phase, although it is also possible that we only have the canonical form since in the Cooks method, we form the proton-bound dimers via electrospray of an aqueous solution, which should favor guanine in the canonical form. We also examined O6-methylguanine (OMG), a highly mutagenic damaged base that arises from the alkylation of guanine. Our calculations indicate that OMG may exist as both the “N9” (canonical) and “N7” (proton on N7 rather than N9) tautomers in the gas phase, as both are calculated to be within 3 kcal mol−1 in energy. We have bracketed the acidity and proton affinity of OMG, which were previously unknown. The more acidic site of OMG has a ΔHacid value of 338 ± 3 kcal mol−1 (ΔGacid = 331 ± 3 kcal mol−1). We have also bracketed the less acidic site (ΔHacid = 362 ± 3 kcal mol−1, ΔGacid = 355 ± 3 kcal mol−1) and the PA (229 ± 4 kcal mol−1 (GB = 222 ± 4 kcal mol−1)). We confirmed these results through Cooks kinetic method measurements as well. Our ultimate goal is to understand the intrinsic reactivity of nucleobases; gas phase acidic and basic properties are of interest for chemical reasons and also possibly for biological purposes, since biological media can be quite nonpolar. We find that OMG is considerably less acidic at N9 than adenine and guanine and less basic at O6 than guanine; the biological implications of these differences are discussed.
Co-reporter:Wickliffe O. Wepukhulu, Vanessa L. Smiley, Bhargavi Vemulapalli, Jeffrey A. Smiley, Linda M. Phillips and Jeehiun K. Lee
Organic & Biomolecular Chemistry 2008 vol. 6(Issue 24) pp:4533-4541
Publication Date(Web):30 Oct 2008
DOI:10.1039/B812979G
Orotidine-5′-monophosphate decarboxylase (OMP decarboxylase, ODCase) catalyzes the decarboxylation of orotidine-5′-monophosphate (OMP) to uridine-5′-monophosphate (UMP). Despite extensive enzymological, structural, and computational studies, the mechanism of ODCase remains incompletely characterized. Herein, carbon kinetic isotope effects were measured for both the natural abundance substrate and a substrate mixture synthesized for the purpose of carrying out the remote double label isotope effect procedure, with O2 of the substrate as the remote position. The carbon kinetic isotope effect on enzymatic decarboxylation of this substrate mix was measured to be 1.0199 ± 0.0007, compared to the value of 1.0289 ± 0.0009 for natural abundance OMP, revealing an 18O2 isotope effect of 0.991 ± 0.001. This value equates to an intrinsic isotope effect of approximately 0.983, using a calculated commitment factor derived from previous isotope effect data. The measured 18O2 isotope effect requires a mechanism with one or more enzymatic processes, including binding and/or chemistry, that contribute to this substantial inverse isotope effect. 18O2 kinetic isotope effects were calculated for four proposed mechanisms: decarboxylation preceded by proton transfer to 1) O2; 2) O4; and 3) C5; and 4) decarboxylation without a preceding protonation step. A mechanism involving no pre-decarboxylation step does not appear to have any steps with the necessary substantial inverse 18O2 effect, thus calling into question any mechanism involving simple direct decarboxylation. Protonation at O2, O4, or C5 are all calculated to proceed with inverse 18O2 effects, and could contribute to the experimentally measured value. Recent crystal structures indicate that O2 of the substrate appears to be involved in an intricate bonding arrangement involving the substrate phosphoryl group, an enzyme Gln side chain, and a bound water molecule; this interaction likely contributes to the observed isotope effect.
Co-reporter:Wickliffe O. Wepukhulu, Vanessa L. Smiley, Bhargavi Vemulapalli, Jeffrey A. Smiley, Linda M. Phillips and Jeehiun K. Lee
Organic & Biomolecular Chemistry 2008 - vol. 6(Issue 24) pp:NaN4541-4541
Publication Date(Web):2008/10/30
DOI:10.1039/B812979G
Orotidine-5′-monophosphate decarboxylase (OMP decarboxylase, ODCase) catalyzes the decarboxylation of orotidine-5′-monophosphate (OMP) to uridine-5′-monophosphate (UMP). Despite extensive enzymological, structural, and computational studies, the mechanism of ODCase remains incompletely characterized. Herein, carbon kinetic isotope effects were measured for both the natural abundance substrate and a substrate mixture synthesized for the purpose of carrying out the remote double label isotope effect procedure, with O2 of the substrate as the remote position. The carbon kinetic isotope effect on enzymatic decarboxylation of this substrate mix was measured to be 1.0199 ± 0.0007, compared to the value of 1.0289 ± 0.0009 for natural abundance OMP, revealing an 18O2 isotope effect of 0.991 ± 0.001. This value equates to an intrinsic isotope effect of approximately 0.983, using a calculated commitment factor derived from previous isotope effect data. The measured 18O2 isotope effect requires a mechanism with one or more enzymatic processes, including binding and/or chemistry, that contribute to this substantial inverse isotope effect. 18O2 kinetic isotope effects were calculated for four proposed mechanisms: decarboxylation preceded by proton transfer to 1) O2; 2) O4; and 3) C5; and 4) decarboxylation without a preceding protonation step. A mechanism involving no pre-decarboxylation step does not appear to have any steps with the necessary substantial inverse 18O2 effect, thus calling into question any mechanism involving simple direct decarboxylation. Protonation at O2, O4, or C5 are all calculated to proceed with inverse 18O2 effects, and could contribute to the experimentally measured value. Recent crystal structures indicate that O2 of the substrate appears to be involved in an intricate bonding arrangement involving the substrate phosphoryl group, an enzyme Gln side chain, and a bound water molecule; this interaction likely contributes to the observed isotope effect.
.jpg)







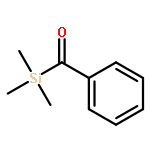
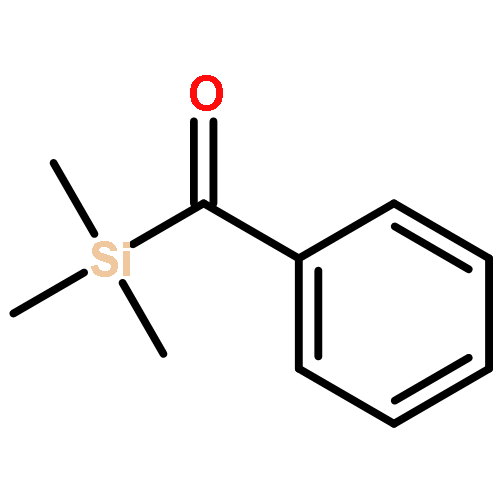
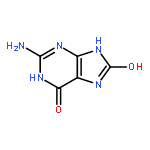

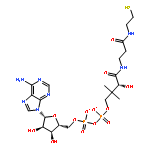

























![7-Methyl-3H-imidazo[4,5-b]pyridine](http://img.cochemist.com/ccimg/27600/27582-20-3.png)
![7-Methyl-3H-imidazo[4,5-b]pyridine](http://img.cochemist.com/ccimg/27600/27582-20-3_b.png)



![7-Nitro-3H-imidazo[4,5-b]pyridine 4-oxide](http://img.cochemist.com/ccimg/14500/14432-11-2.png)
![7-Nitro-3H-imidazo[4,5-b]pyridine 4-oxide](http://img.cochemist.com/ccimg/14500/14432-11-2_b.png)


![3H-Imidazo[4,5-b]pyridin-7-amine](http://img.cochemist.com/ccimg/6800/6703-44-2.png)
![3H-Imidazo[4,5-b]pyridin-7-amine](http://img.cochemist.com/ccimg/6800/6703-44-2_b.png)







![1-Phenyl-1H-benzo[d][1,2,3]triazole](http://img.cochemist.com/ccimg/900/883-39-6.png)
![1-Phenyl-1H-benzo[d][1,2,3]triazole](http://img.cochemist.com/ccimg/900/883-39-6_b.png)
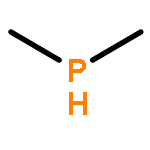
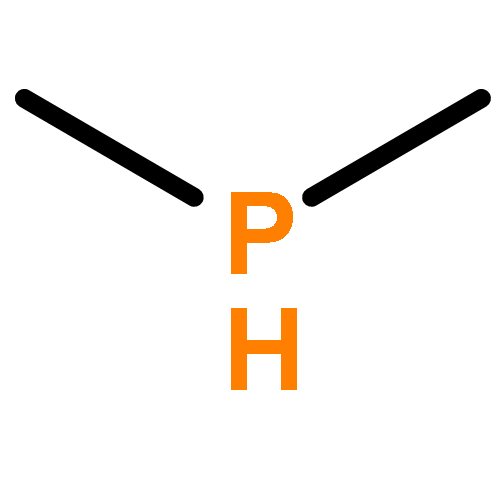
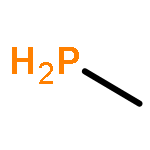
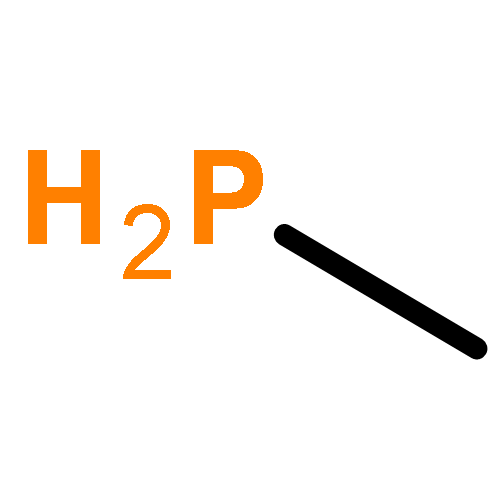


![2l5-1,3,2-Diazaphosphorin-2-amine,2-[(1,1-dimethylethyl)imino]-N,N-diethylhexahydro-1,3-dimethyl-](http://img.cochemist.com/ccimg/98100/98015-45-3.png)
![2l5-1,3,2-Diazaphosphorin-2-amine,2-[(1,1-dimethylethyl)imino]-N,N-diethylhexahydro-1,3-dimethyl-](http://img.cochemist.com/ccimg/98100/98015-45-3_b.png)