Co-reporter:Zhimin Xie, Dongliang Chai, Youshan Wang, and Huifeng Tan
The Journal of Physical Chemistry B 2016 Volume 120(Issue 45) pp:11834-11844
Publication Date(Web):October 21, 2016
DOI:10.1021/acs.jpcb.6b06457
Effective potentials are of great importance for coarse-grained (CG) simulations, which can be obtained by the structure-based iterative Boltzmann inversion (IBI) method. However, the standard IBI method is incapable of maintaining the mechanical and thermodynamic properties of the CG model in agreement with those of the all-atom model. Unlike the existing techniques, such as introducing friction force as the dissipative force to reduce the superatom motion while keeping the conservative force arising from the CG potential intact, we directly modified the standard IBI nonbonded potential by adding an empirical function. According to an analysis of the dissipative particle dynamics, the additional function did compensate for the friction reduction of the standard IBI CG model. In this work, the thermal fluctuation information from the nonbonded radial distribution function was incorporated into the additional empirical function. As an illustration of the new CG force fields, we presented simulations of the stress–strain relation and thermodynamic properties in terms of cis-polyisoprene and compared the statistical structure information of the superatoms with those of the IBI CG model and the all-atom model. It should be emphasized that the additional empirical function contributed to compensating for the friction reduction, irrespective of the functional form it took. In this sense, the proposed method was easily operable.
Co-reporter:Dongliang Chai, Zhimin Xie, Youshan Wang, Li Liu, and Young-Jin Yum
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 20) pp:17974
Publication Date(Web):October 2, 2014
DOI:10.1021/am504799m
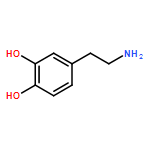
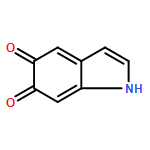
Dopamine, as a universal material for surface treatment, can effectively improve the surface performance of aramid fibers. However, directly processing the surface of aramid fibers using dopamine currently incurs a high cost. To seek dopamine substitutes, one must first explore the adhesion mechanism responsible for binding the dopamine to the surface of the fiber. In this study, we construct an all-atomic molecular dynamics model of an aramid fiber before and after surface modification using dopamine. A force field based on condensed-phase optimized molecular potentials for atomistic simulation studies (COMPASS) is used. Using it, we analyze the surface adhesion mechanism of polydopamines aggregated by 21 kinds of molecular structures typically found on the surface of aramid fibers. The results show that a clear and smooth interface is formed between the polydopamine nanofilm layer and the surface of the aramid fiber. The high atomic density of the polydopamine in the small interface region is found to be conducive to noncovalent bonds of polydopamines with the surface of the aramid fiber. In addition, we investigate the works of adhesion of the 21 molecular structures typically found on the surface of aramid fibers. The results suggest that the work of adhesion of 5,6-indolequinone is the highest, followed by annular eumelanin molecules with annular planar structure. Straight-chain shaped dimers proved to be the molecules with the highest adhesion ability of the dihydroxyindole chain oligomers. Therefore, there is reason to suppose that more molecular structures (as above) can be formed by processing the surface of aramid fibers using dopamine by controlling the processing conditions. These molecular structures help improve the adhesion ability of the dopamine on the surface of the aramid fiber. Additionally, if these polydopamine molecules with high adhesion ability can be synthesized on a large scale, then new surface-processing materials are possible.Keywords: molecular dynamics; poly(p-phenylene terephthalamide); polydopamine; surface energy; work of adhesion